
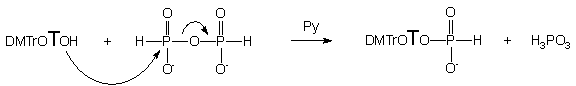
| 119 | 120 |
|
Schemat 17
Pomimo długiego czasu reakcji zdecydowałem się jednak podjąć prace nad kwasem pirofosfonowym jako czynnikiem fosfonylującym, gdyż reakcje fosfonylacji z jego użyciem zachodzą wyjątkowo czysto i bez produktów ubocznych. Reagent ten wykorzystywałem do syntezy H-fosfonianu 5'-O-(4,4'-dimetoksytrytylo)-tymidyny 121. W tym celu do roztworu kwasu pirofosfonowego w pirydynie (5 ekw.), otrzymanego w reakcji kwasu fosforawego i czynnika kondensującego (chlorek piwaloilu lub NEP-Cl), dodawałem roztwór 5'-O-(4,4'-dimetoksytrytylo)-tymidyny 119 (1 ekw.) i pozostawiałem na noc. Analiza TLC i 31P NMR nie wykazywała powstawania jakichkolwiek produktów ubocznych*. Czysty związek 121 izolowałem z wydajnością powyżej 80%.
Można się spodziewać, że adaptacja tego systemu do fosfonylacji oligonukleotydu związanego z podłożem stałym wymagać będzie użycia zdecydowanie wyższych nadmiarów reagenta. W takiej sytuacji zasady azotowe będą eksponowane na działanie dużych nadmiarów molowych czynnika fosfonylującego, co stwarza niebezpieczeństwo zachodzenia niepożądanych reakcji ubocznych, szczególnie na O-4 tyminy i O-6 guaniny. Podjąłem więc badania nad możliwością zachodzenia takich reakcji. Substratami były odpowiednio zablokowana deoksytymidyna [(5'-O-(4,4'-dimetoksytrytylo), 3'-O-benzolilo] i deoksyguanozyna [5'-O-(4,4'-dimetoksytrytylo), 3'-O-benzoilo, N2-izoburtyrylo], które traktowano 30-krotnym nadmiarem kwasu pirofosfonowego. Jako metodę analityczną zastosowałem 31P NMR, gdyż w przypadku pojawienia się produktów O-fosfonylacji, spodziewane monoestry H-fosfonianowe, przy dużej czułości aparatu jakim dysponowałem, powinny być łatwe do wykrycia. W widmach 31P NMR mieszanin pochodnych guanozyny i tymidyny traktowanych przez kilka dni (72 h) kwasem pirofosfonowym nie stwierdziłem powstawania potencjalnych produktów fosfonylacji.
Te wyniki oraz wcześniej opisane właściwości fosfonylujące kwasu pirofosfonowego w pirydynie były podstawą podjęcia dalszych badań, ale już na oligomerach przyłączonych do podłoża.
W badaniach nad transestryfikacją diestrów H-fosfonianowych (opisanych w dalszej części pracy) stwierdziłem, że w kontrolowanych warunkach H-fosfonianowe diestry arylowo - nukleozydowe bardzo łatwo ulegają chemoselektywnej transestryfikacji alkoholami do diestrów alkilowo - nukleozydowych. Te wyniki zachęciły mnie do podjęcia prac nad fosforynem difenylowym jako czynnikiem wprowadzającym resztę H-fosfonianową do nukleozydów. Reagent ten nie wymaga stosowania czynników kondensujących, gdyż aktywność centrum fosforowego uwarunkowana jest wyciągającymi elektrony podstawnikami arylowymi. Czynnikiem potencjalnie limitującym zastosowania fosforynu difenylowego może być fakt, że produktem pierwszej reakcji transestryfikacji jest H-fosfonian alkilowo - arylowy, który może ulegać dalszej reakcji transestryfikacji nadmiarem alkoholu, prowadząc do H-fosfonianu dialkilowego.
Podjąłem więc prace nad zastosowaniem fosforynu difenylowego do fosfonylacji nukleozydów, których celem było określenie czynników warunkujących szybkość i chemoselektywność reakcji fosfonylacji nukleozydów i dalszych reakcji prowadzących do H-fosfonianów nukleozydów jako produktów docelowych (Schemat 18).
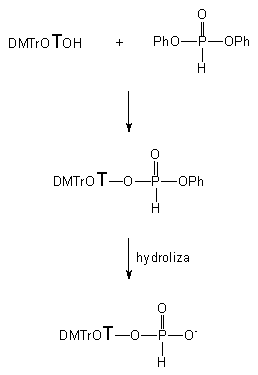
Schemat 18
W pierwszej kolejności badałem proces chemoselektywności fosfonylacji w zależności od stosunków molowych substratów tj. 3'-OH nukleozydu i fosforynu difenylowego. Reakcje prowadzone były w pirydynie przy stałym stężeniu nukleozydu (1 mmol/10 ml roztworu) a jako metodę analityczną stosowałem ponownie 31P NMR (monitorowanie produktów zawierających atom fosforu) i TLC (monitorowanie substratu nukleozydowego i nukleotydowego produktu fosfonylacji). Zgodnie z oczekiwaniami, chemoselektywność reakcji silnie zależała od nadmiaru zastosowanego fosforynu difenylowego. I tak, użycie 0,5 ekw. fosforynu prowadziło do powstawania na pierwszym etapie H-fosfonianowego diestru fenylowo - nukleozydowego, który ulegał dalszej transestryfikacji nadmiarem nukleozydu, tworząc wolno (12 godzin) symetryczny H-fosfonian 3'-3' dinukleozydowy jako jedyny produkt obserwowany w mieszaninie reakcyjnej. Zastosowanie kolejno 1, 3 i 5 ekw. czynnika fosfonylującego zasadniczo zmieniło rozkład produktów, gdyż po wyczerpaniu nukleozydu obserwowano w mieszaninach poreakcyjnych odpowiednio 21%, 4% i 0% 3'-3' symetrycznego związku. W ostatnim przypadku pożądany diester fenylowo - nukleozydowy był jedynym wykrywalnym produktem nukleotydowym. Reakcje zachodziły szybko i były zakończone w czasie zaledwie kilku minut (5 - 10 min. w zależności od nadmiaru czynnika fosfonylującego). Prowadzona w następnym etapie hydroliza wiązania arylofosforoestrowego w otrzymanych H-fosfonianach arylowo - nukleozydowych zachodziła także szybko (5 min.) i z całkowitą chemoselektywnością. Produkty fosfonylacji - H-fosfoniany nukleozydów (zarówno serii rybo- i deoksyrybo-) izolowano z wydajnościami powyżej 90%. Powyższe prace doprowadziły do nowej, szybkiej i wydajnej metody otrzymywania H-fosfonianów nukleozydów, nie wymagającej stosowania czynników kondensujących, a wykorzystującej wyjątkową łatwość transestryfikacji arylowych diestrów H-fosfonianów.
Ze względów opisanych wcześniej, także i w tym przypadku całkowicie zablokowane nukleozydy poddano działaniu 30-krotnego nadmiaru molowego fosforynu difenylowego. Monitorowanie tak przygotowanej mieszaniny metodą 31P NMR nie wykazało powstawania potencjalnych produktów reakcji czynnika fosfonylującego z zasadami azotowymi.
Fosforyn difenylowy w porównaniu z kwasem pirofosfonowym okazał się równie skutecznym i jednocześnie o wiele szybciej działającym czynnikiem fosfonylującym. Fakt, że reagent ten na pierwszym etapie fosfonylacji tworzy H-fosfonianowe diestry alkilowo - arylowe, w sprzyjających warunkach zdolne do dalszych transestryfikacji z nieprzereagowanym ligandem hydroksylowym, zmusza do bardzo ostrożnej weryfikacji jego stosowania do fosfonylacji oligonukleotydów związanych z podłożem stałym. Dopiero wyniki tych doświadczeń pozwolą na ocenę przydatności fosforynu difenylowego jako reagenta wprowadzającego resztę H-fosfonianową na koniec 5'-oligonukleotydu.
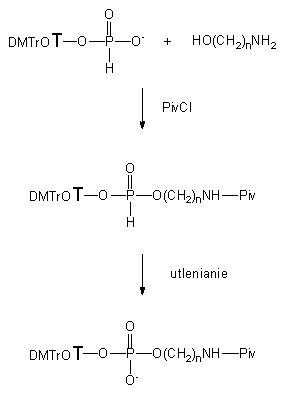
Schemat 19
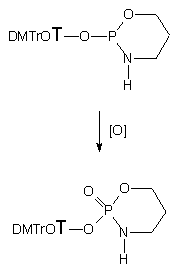
W reakcji 3'-H-fosfonianu 5'-O-(4,4'-dimetoksytrytylo)-tymidyny 121 z aminoalkoholami 124a, b, e* w warunkach standardowych [aktywacja nukleotydu chlorkiem piwaloilu (2,5 ekw.) w obecności aminoalkoholu (1,1 ekw.) w pirydynie]120 powstają produkty, dające w 31P NMR sygnały charakterystyczne dla H-fosfonianowych diestrów alkilowych. Reakcja jest szybka, czysta i chemoselektywna (nie stwierdzono obecności związków z wiązaniem P-N). Próby izolacji produktów reakcji zakończyły się jednak niepowodzeniem ze względu na ich nietrwałość podczas przerobu i chromatografii. W celu ich identyfikacji utleniłem je (3% roztwór jodu w pirydynie zawierającej 10% wody) i oczyściłem za pomocą chromatografii kolumnowej na żelu krzemionkowym. Analiza widma 1H-NMR i MS potwierdziła, że są to fosforany nukleozydowo - aminoalkilowe. Ponadto okazało się, że funkcja aminowa tych związków jest acylowana grupą piwaloilową. Acylowanie zachodzić może w różnych fazach reakcji, nie podjąłem jednak badań mających na celu dokładne przebadanie tego procesu, gdyż niezależnie od momentu, w którym następuje acylowanie, otrzymuje się produkty typu 125, których dalsze aplikacje są raczej ograniczone ze względu na dużą trwałość wiązania karbamidowego. Przebieg powyższych reakcji przedstawiony jest na Schemacie 19.
H-Fosfonian 121 w pirydynie poddałem działaniu 2,5 ekw. chlorku piwaloilu generując fosforyn nukleozydowo - dipiwaloilowy 127, który następnie traktowałem aminoalkoholami 124a - e (Schemat 20). Mając na uwadze na mnogość centr reaktywnych w preaktywowanym nukleotydzie typu 127 i aminoalkoholu oraz liczbę kombinacji z tym związanych, można się spodziewać powstawania wielu produktów reakcji. Analizując mieszaniny reakcyjne metodą 31P NMR można było zauważyć, że charakter i dystrybucja produktów zależy od rodzaju użytego aminoalkoholu.
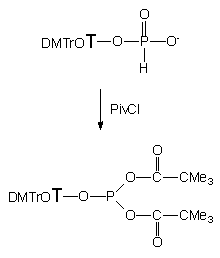 127
127
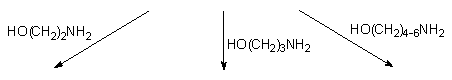
 |
128a
129a
130
|
 |
128b
129b |
mieszanina triestrów fosforynowych |
Schemat 20
Tak więc, fosforyn nukleozydu 127 w reakcji z aminoetanolem (124a) prowadził do produktu rezonującego przy ok. 138 ppm. Sądząc z położenia sygnału oraz jego multipletowości można postulować, że jest to cykliczny amidofosforyn nukleozydu typu 128a. Powstawanie takiego produktu jest możliwe w sekwencji reakcji, w których na pierwszym etapie następuje atak nukleofilowy na centrum fosforowe z odejściem jednej reszty piwaloilowej i z wytworzeniem liniowego estru, 2-aminoetylofosforynu lub też N-(2-hydroksyetylo)amidofosforynu nukleozydowo - piwaloilowego. Na następnym etapie wciąż aktywowane (grupą piwaloilową) centrum fosforowe atakowane jest wewnątrzcząsteczkowo przez nukleofil (-OH lub -NH2) z pozycji b, co prowadzi do zamknięcia pierścienia, z wytworzeniem cyklicznego amidofosforynu nukleozydu 128a. Mimo podjętych prób, ze względu na szybkość zachodzących reakcji, nie udało mi się ustalić, który nukleofil (-OH czy -NH2) atakuje centrum fosforowe jako pierwszy. Próby izolacji fosfolidyny 128a nie powiodły się, gdyż w trakcie przerobu związek ten był nietrwały i rozkładał się niespecyficznie do kilku produktów. Spodziewając się, że analog fosforanowy będzie trwalszy, podjąłem próbę utlenienia związku 128a jodem wobec niewielkiego (15 ekw.), ale koniecznego nadmiaru wody. W tych warunkach powstawał jeden produkt rezonujący przy 8,2 ppm. Został on wyizolowany i na podstawie analiz 1H, 31P NMR i MS jego struktura została określona jako N-(2-hydroksyetylo)amidofosforan nukleozydu 130. Można przyjąć, że otrzymany związek 130 jest produktem hydrolizy cyklicznego amidofosforanu 129a. Jeżeli jest tak faktycznie, to jest to produkt niespodziewany, gdyż na podstawie wcześniej opisanych prac nad hydrolizą w warunkach zasadowych analogicznych pięcioczłonowych układów heterocyklicznych (pochodne N-alkilowe i N-fenylowe)121 należało oczekiwać otwierania pierścienia z rozerwaniem wiązania P-N, z wytworzeniem fosforodiestru nuleozydowo - aminoalkilowego.
Ponieważ w trakcie utleniania, ze względu na szybkość procesu, nie rejestrowałem domniemanego cyklicznego amidofosforanu 129a, wygenerowałem go, aktywując oczyszczony produkt 130 chlorkiem arylosulfonowym (TPS-Cl). W tej reakcji, zachodzącej szybko (< 3 min.) i czysto, powstawał jeden produkt - cykliczny amidofosforan nukleozydu 129a. Mimo, że związek ten nie został wyizolowany, to jego przesunięcie chemiczne (31P NMR) i multipletowość sygnałów w widmie sprzężonym mocno sugerują proponowaną strukturę. Dodanie wody do mieszaniny reakcyjnej powoduje natychmiastową hydrolizę 129a do wyjściowego N-(2-hydroksyetylo)amidofosforanu nukleozydu 130. To doświadczenie można traktować jako pozytywną weryfikację wcześniej postulowanego szeregu reakcji utleniania cyklicznego amidofosforynu nukleozydu 128a do cyklicznego amidofosforanu 129a i dalej w łagodnych warunkach zasadowych jego hydrolizę do finalnego produktu 130.
Na tym etapie prac nie potrafię wyjaśnić różnic w chemoselektywności otwierania powyżej opisanych pierścieni heterocyklicznych, gdyż ilość danych doświadczalnych, którymi dysponuję jest zbyt skąpa. W czasie realizacji tej rozprawy takich prób nie podjąłem, jednakże sądzę, że zastosowanie N-alkilo- lub N-aryloaminoetanoli w analogicznych reakcjach mogłoby dostarczyć danych, które pozwoliłyby lepiej poznać i zdefiniować czynniki decydujące o chemoselektywności hydrolizy w warunkach zasadowych omawianych związków.
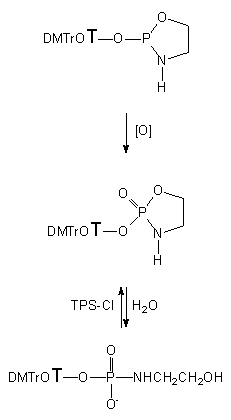
3-Aminopropan-1-ol (124b) na pierwszych etapach reakcji reaguje z dipiwaloilofosforynem nukleozydu 127 podobnie jak 2-aminoetanol, tworząc cykliczny amidofosforyn nukleozydu 128b (31P NMR). Reakcja cyklizacji zachodzi bardzo łatwo a jej siłą napędową jest tendencja do wewnątrzcząsteczkowego ataku nukleofila (-OH lub -NH2) z pozycji g na centrum fosforowe. Utlenianie 128b w obecności 15 ekw. H2O prowadzi do trwałego cyklicznego amidofosforanu nukleozydu 129b - 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-2-okso-1,3,2-oksazaperhydrofosforinu, który wyizolowano i scharakteryzowano metodami spektroskopowymi (1H, 31P NMR i HRMS). Mając na uwadze fakt, że reakcje cyklizacji i utleniania zachodzą szybko i wydajnie, a cykliczny amidofosforan nukleozydu 129b jest związkiem trwałym, można traktować powyższy cykl reakcji jako dogodną metodę syntezy pochodnych nukleotydów typu 129b.
Wyższe aminoalkohole 124c - e w reakcji z fosforynem 127 prowadziły do mieszaniny wielu produktów, których nie izolowano ze względu na ich nietrwałość. Jest bardzo prawdopodobne, że w tych przypadkach otrzymuje się różnego typu (estry lub/i amidy) i różnie podstawione (mono i bis) fosforyny nukleozydów co może być konsekwencją mnogości centr aktywnych substratów. W odróżnieniu od 2-aminoetanolu i 3-aminopropan-1-olu, wyższe homologi nie tworzą struktur cyklicznych typu 128, które w pierwszych dwóch przypadkach decydowały o kierunku dalszych reakcji.
W celu weryfikacji poprawności tych założeń, 2-aminoetanol
124a
traktowałem równomolową ilością bezwodnika trifluorooctowego (TFAA) w pirydynie.
Otrzymaną mieszaninę wykorzystałem bezpośrednio do kondensacji z monoestrem
H-fosfonianowym 121wobec chlorku piwaloilu. Po utlenieniu
w standardowych warunkach otrzymałem fosforan
131a
(31P NMR). Grupę trifluoroacetylową
usunąłem poprzez traktowanie stężonym roztworem amoniaku (Schemat 21).
Analogiczne reakcje doprowadziły do otrzymania diestrów fosforanowych również
dla innych aminoalkoholi (związki 131b - e).
Po zakończeniu cyklu reakcji pożądane produkty izolowałem za pomocą chromatografii
na żelu krzemionkowym z wydajnościami powyżej 80%.
|
121 |
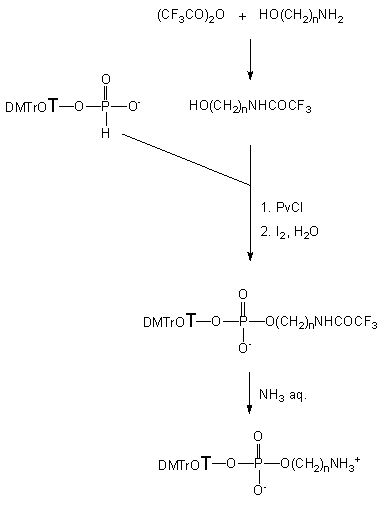 |
124a - e 131a -
e
132a - e |
Schemat 21
Podejście z preacylacją aminoalkoholu można traktować jako uniwersalną metodę syntezy aminoalkilofosforanów nukleozydów typu 132, jednakże konieczność stosowania amoniaku w celu odsłonięcia funkcji -NH2 na ostatnim etapie syntezy znacznie ogranicza wykorzystanie tego podejścia do funkcjonalizacji oligonukleotydów związanych z podłożem stałym. Wiąże się to z faktem, że podczas usuwania grupy trójfluorooctowej ugrupowanie karboksyloestrowe wiążące oligomer z podłożem także ulega amonolizie z uwolnieniem sfunkcjonalizowanego oligonukleotydu do roztworu. W konsekwencji, w tym podejściu, przy stosowaniu powszechnie używanych podłoży stałych, realizacja ostatniego etapu Wariantu I (przyłączenie grupy reporterowej do oligonukleotydu zakotwiczonego do podłoża) nie jest możliwa.
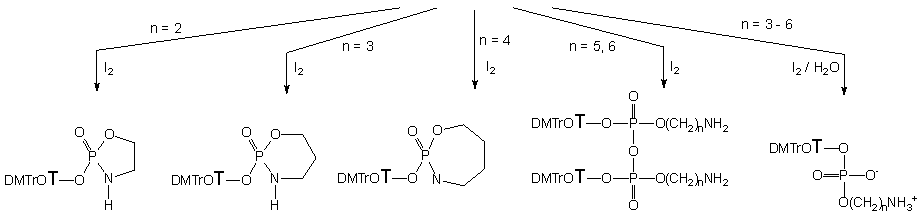
Reakcje H-fosfonianu nukleozydu 121 i aminoalkoholi 124a - e wobec NEP-Cl prowadziły do aminoalkilo-H-fosfonianów nukleozydów typu 134 (Schemat 22). Monitorowanie reakcji za pomocą spektroskopii 31P NMR pozwoliło stwierdzić, że reakcje te zachodzą szybko (<3 min.) i z całkowitą chemoselektywnością, gdyż bez względu na rodzaj użytego aminoalkoholu, jedynymi nukleotydowymi produktami reakcji były H-fosfoniany aminoalkilowo - nukleozydowe typu 134. Próby izolacji tych produktów nie powiodły się, gdyż w czasie przerobu i izolacji diestry typu 134 okazały się niedostatecznie trwałe. Związki te utleniałem oczekując, że ich fosforanowe analogi będzie można wyizolować i w pełni scharakteryzować
Utlenianie H-fosfonianodiestru 134a
jodem wobec dużego nadmiaru wody (300 ekw.) prowadziło do otrzymania produktu,
którego przesunięcie chemiczne w widmie 31P NMR było identyczne
z otrzymanym wcześniej N-(2-hydroksyetylo)amidofosforanem nukleozydu 130.
Identyczność otrzymanego produktu ze związkiem 130 została
potwierdzona także innymi metodami analitycznymi (1H NMR, TLC,
testy chemiczne - reakcja z TPS-Cl - odtworzenie cyklicznego amidofosforanu).
Można przypuszczać, że związek ten jest końcowym produktem cyklu reakcji
zapoczątkowanym wytworzeniem jodofosforanu jako pierwszego produktu utleniania
diestru H-fosfonianowego 134. Po to, by wyjaśnić tę sprawę,
przeprowadziłem utlenianie diestru 134 w warunkach
bezwodnych. Jedynym produktem obserwowanym w widmie 31P
NMR był poznany już wcześniej cykliczny amidofosforan 129a.
Dodanie wody do mieszaniny reakcyjnej powodowało szybką hydrolizę 129a
do trwałego amidofosforanu nukleozydu 130 jako jedynego produktu.
Tak więc, reakcja wewnątrzcząsteczkowej cyklizacji do produktu pośredniego
129a
zachodzi z chwilą powstania jodofosforanu na pierwszym etapie utleniania
bez względu na nadmiar konkurencyjnego nukleofila (H2O),
a finalny produkt
130 jest produktem
następczej, chemoselektywnej reakcji hydrolizy amidofosforanu nukleozydu
129a.
|
121 |
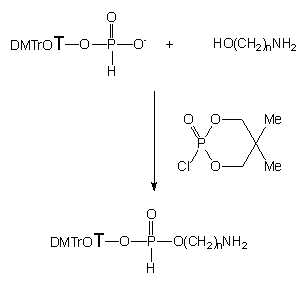 |
124a
-
e
133 |
|
|
|
|
|
|
 |
|
|
Schemat 22
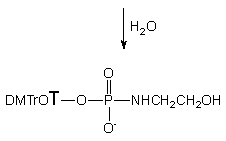
Typ produktów utleniania jodem 3-aminopropylo-H-fosfonianu nukleozydu 134b zależał od ilości wody obecnej w medium reakcyjnym. I tak, utlenianie 134b w warunkach bezwodnych prowadziło ilościowo do trwałego cyklicznego amidofosforanu nukleozydu 129b. Reakcja ta zachodzi wyjątkowo czysto i można ją polecać jako dogodną metodę szybkiej syntezy cyklicznych pochodnych nukleotydów typu 129b z H-fosfonianów nukleozydów typu 121 jako łatwo dostępnych substratów. Utlenianie 134b wobec niewielkiego nadmiaru wody prowadziło do powstawania dwóch produktów - opisanego wcześniej cyklicznego amidofosforanu 129b oraz symetrycznego pirofosforanu 135b. Z dużą dozą pewności można postulować, że ten ostatni związek powstaje w reakcji niezhydrolizowanego jodofosforanu (pierwszego produktu utlenienia) z produktem już zhydrolizowanym tj. 132b. Dodatkowym potwierdzeniem takiego rozumowania było doświadczenie, w którym utleniano diester H-fosfonianowy 134b w bezwodnych warunkach wobec fosforodiestru 132b. Jedynym produktem w tej reakcji był stosunkowo trwały pirofosforan 135b.
Stosunek cyklicznego produktu 129b i pirofosforanu 135b zmieniał się w zależności od ilości wprowadzanej wody w czasie utleniania, przy czym wobec większych nadmiarów H2O zaczął się pojawiać pożądany fosforodiester 132b. Jeżeli reakcję utleniania 134b przeprowadzano przy dużych nadmiarach (300 ekw.) wody, to 3-aminopropylofosforodiester nukleozydu 132b był jedynym otrzymanym produktem. Tak więc nadmiar wody jako zewnętrznego nukleofila całkowicie zapobiega zachodzeniu konkurencyjnych reakcji cyklizacji czy dimeryzacji. Po izolacji, strukturę produktu 132b ustalono rutynowymi metodami spektralnymi (1H, 31P NMR, HRMS) i chromatograficznymi (TLC).
H-Fosfonianodiester 134c z grupą 4-aminobutylową, utleniany w warunkach bezwodnych także tworzył siedmioczłonowy cykliczny amidofosforan nukleozydu 129c. Manipulowanie ilością wody podczas utleniania pozwoliło, podobnie jak w przypadku pochodnej 3-aminopropylowej 134b, wykluczyć cyklizację czy dimeryzację i w konsekwencji pożądany produkt finalny 132c był otrzymany z wysoką wydajnością.
W przypadku pochodnych wyższych homologów aminoalkoholi podczas utleniania nie obserwowano powstawania struktur cyklicznych, a odpowiednie nadmiary wody pozwoliły na ukierunkowanie reakcji na tworzenie wyłącznie aminoalkilofosforanoamidów nukleozydów 132d - e.
Opisane procedury można traktować jako uniwersalną metodę syntezy aminoalkilowych fosforanów nukleozydów typu 132. Wyjątek stanowią pochodne 2-aminoetylowe, w których w przypadku aktywacji centrum fosforowego (tworzenie jodofosforanów) tendencja do wewnątrzcząsteczkowej substytucji jest tak silna, że pomimo stosowania dużych nadmiarów zewnętrznego nukleofila nie udało się zapobiegać cyklizacji.
Opracowana metoda, angażująca proste substraty i procedury spełnia kryteria funkcjonalizacji oligonukleotydów wg Wariantu I, a jej efektywność będzie weryfikowana (w dalszej części rozprawy) na oligonukleotydach związanych z podłożem stałym.
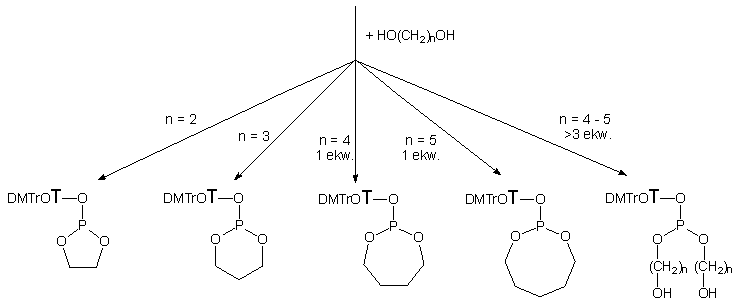
I tak, glikol etylenowy (136a) i
propanodiol-1,3 (136b) bez względu na nadmiar molowy, w reakcji
z bis-acylofosforynem 127 tworzyły produkty cykliczne, odpowiednio
2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-1,3,2-dioksafosfolan
(137a) i 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-1,3,2-dioksafosfinan
(137b). Diole 136c i 136d dodane
w niewielkim (1,5 ekw.) nadmiarze do związku 127 także tworzyły
cykliczne fosforyny nukleozydu 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-1,3,2-dioksafosfepan
(137c) i 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-1,3,2-dioksafosfokan
(137d) jako główne produkty. Zwiększanie nadmiaru dioli 136c
i 136d prowadziło do powstawania liniowych
di(hydroksyalkilowych) fosforynów nukleozydów 138c
i 138d, które przy nadmiarze powyżej 3
ekw. były głównymi produktami reakcji. Z tych doświadczeń można wnioskować,
że w przypadku dioli 136a i 136b, tendencja
do tworzenia pierścieni pięcio- lub sześcioczłonowych
jest tak silna, że bez względu na nadmiar diolu, cykliczne fosforyny 137a
i 137b są jedynymi produktami. W przypadku
dioli 136c i 136d
reakcje cyklizacji zachodzą jedynie w sprzyjających warunkach i konkurują
z produktami międzycząsteczkowej substytucji prowadzącej do liniowych triestrów
fosforawych
138c i 138d.
|
121
127 |
|
|
|
|
|
|
|
|
|
|
|
|
 |
||||
|
|
Schemat 22
Próby oczyszczania produktów cyklicznych 137b - d nie powiodły się, gdyż były one nietrwałe w warunkach izolacji. Cykliczny fosforyn nukleozydu 137a okazał się natomiast dostatecznie trwały i po izolacji został scharakteryzowany metodami spektralnymi (1H oraz 31P NMR).
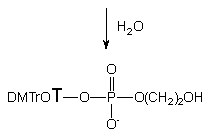
Utlenianie cyklicznych fosforynów 137b - d jodem wobec wody prowadziło do otrzymania trwałych cyklicznych fosforanów 139b - d, które wyizolowano, a ich struktury potwierdzono metodami spektralnymi. W tych samych warunkach produktem utleniania fosfolanu 137a był fosforan 5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-ylowo - 2-hydroksyetylowy 141a, który jak można przypuszczać, był produktem hydrolizy powstałego na pierwszym etapie cyklicznego fosforotriestru 139a {2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-2-okso-1,3,2-dioksafosfolanu}. Taki sposób powstawania fosforodiestru 141a został dowiedziony przez zastosowanie niewielkiego (1,5 ekw.) nadmiaru wody podczas utleniania. W tych warunkach jedynym obserwowanym (31P NMR) produktem był cykliczny fosfolan 139a, który po dodaniu wody szybko hydrolizował z otwarciem pierścienia do 2-hydroksyetylofosforanu nukleozydu 141a. Aktywowanie (TPS-Cl) centrum fosforowego diestru 141a powodowało natychmiastową cyklizację do 139a. Z doświadczeń tych wynika, że fosforodiester 2-hydroksyetylowo - nukleozydowy 141a jest związkiem podatnym na cyklizację, a produkt tej reakcji łatwo hydrolizuje do wyjściowego liniowego estru 141a.
Podsumowując, jedynym diolem umożliwiającym otrzymanie diestru typu 141 z wolną grupą hydroksylową w reakcji z preaktywowanym H-fosfonianem typu 127 jest glikol etylenowy. W przypadku pozostałych badanych dioli 136b - d otrzymuje się trwałe, cykliczne fosforotriestry 139b - d.
Wszystkie badane diole 136a - d
w reakcji z H-fosfonianem nukleozydu 121 aktywowanym cyklicznym
chlorofosforanem 133 prowadziły do pożądanych hydroksyalkilo-H-fosfonianów
nukleozydów typu 142 (Schemat 24). W przypadku dioli
136b
- d, w których funkcje hydroksylowe były
oddzielone co najmniej trzema grupami metylenowymi, spodziewane diestry
liniowe typu 142 były jedynymi produktami reakcji kondensacji.
Natomiast, w mieszaninie poreakcyjnej po kondensacji glikolu etylenowego
i H-fosfonianu 121, diester 142a był produktem
minorowym (31P NMR). Głównymi produktami tej reakcji były: związek
o dP = 23,09 ppm (31P
NMR) oraz 5'-O-dimetoksytrytylotymidyna (TLC).
|
121 |
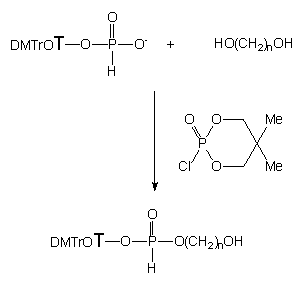 |
136a
- d
133
|
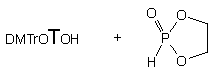 |
|
|
|
Schemat 24
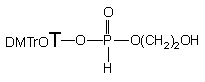
Taki skład mieszaniny poreakcyjnej może świadczyć o występowaniu reakcji następczych, w których substratem jest otrzymany po kondensacji 2-hydroksyetylo-H-fosfonian nukleozydu 142a, i który ulega spontanicznej cyklizacji do fosfolanu 143a z odejściem reszty nukleozydowej. Proporcje ilościowe produktów 142a, 143a i nukleozydu nie zmieniały się w czasie. Dodanie dużego (10 ekw.) namiaru nukleozydu powodowało znaczny wzrost ilości diestru 142a. To ostatnie wskazuje na występowanie równowagi 142a <=> 143a + 119(Schemat 24), która po dodaniu nadmiaru nukleozydu przesuwa się w stronę diestru 142a. Powstawania fosfolanu 143a można było się spodziewać, gdyż wcześniej opisane próby syntezy H-fosfonianu 3'-5' dirybonukleozydowego, w którym 3' nukleozyd miał odsłoniętą wicynalną resztę 2'-OH nie powiodły się ze względu na szybką cyklizację do 2', 3' cyklicznego H-fosfonianu nukleozydu, który w trakcie dalszego przerobu ulegał niespecyficznej hydrolizie do mieszaniny 2' i 3' H-fosfonianów nukleozydu.124
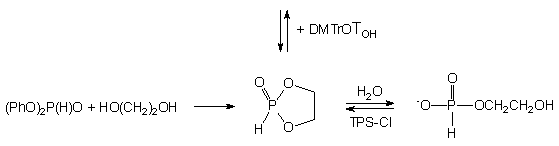
Z powyższych doświadczeń można wnioskować, że centrum fosforowe fosfolanu 143a jest podatne na atak nukleofilowy. Chcąc lepiej poznać właściwości cyklicznego H-fosfonianu 143a, otrzymałem go w reakcji równomolowych ilości glikolu etylenowego z fosforynem difenylowym (Schemat 25).
W pirydynie reakcja ta zachodzi bardzo szybko (z chwilą zmieszania reagentów), a fosfolan 143a jest jedynym obserwowanym produktem (31P NMR). Dodanie nadmiaru wody powoduje szybką jego hydrolizę do H-fosfonianu 2-hydroksyetylowego 144. Po usunięciu wody z medium monoester 144 poddany aktywacji (TPS-Cl) odtwarza ilościowo substrat 143a. Z kolei dodanie etanolu do roztworu 143a w pirydynie powoduje otwieranie pierścienia i przy pięciomolowym nadmiarze całkowitą linearyzację do diestru 145. Usunięcie nadmiaru etanolu z mieszaniny reakcyjnej odtwarza ilościowo substrat 143a. Fosfolan 143a podobnie reaguje z 5'-dimetoksytrytylotymidyną, a ilość 2-hydroksyetylo-H-fosfonianu nukleozydu 142a wzrasta wraz ze zwiększaniem nadmiaru nukleozydu.
Bardzo silna tendencja do wewnątrzcząsteczkowej cyklizacji diestrów 2-hydroksyetylo-H-fosfonianów w zasadzie wyklucza otrzymywanie preparatywne tych związków. Z drugiej strony, przeprowadzanie monoestrów alkilowych (np. nukleozydowych) w ich pochodne 2-hydroksyetylowe diestry może być propozycją dogodnej metody defosfonylacji H-fosfonianów alkilowych (np. nukleozydów bądź oligonukleotydów).
|
|
136a | 143a | 144 |
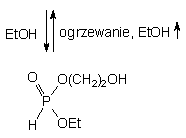
Schemat 25
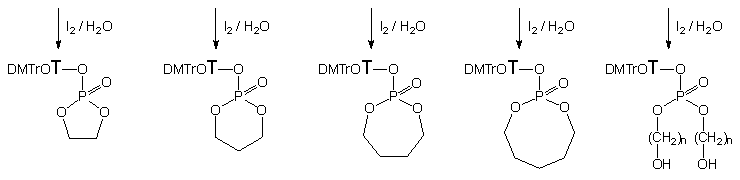
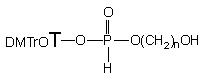
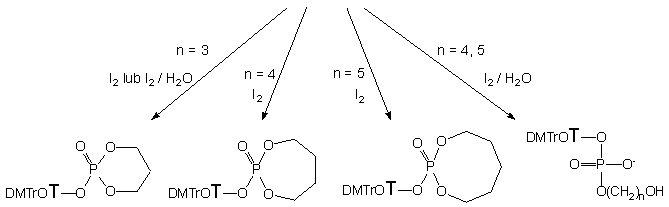
Wyższe hydroksyalkilo-H-fosfoniany nukleozydów 142b - d, otrzymane wg Schematu 24, były utleniane jodem do analogów fosforanowych. W reakcjach tych rodzaj i dystrybucja produktów zależały od warunków utleniania i długości mostka polimetylenowego (Schemat 26). I tak, pochodna 3-hydroksypropylowa 142b, bez względu na nadmiar wody użyty w reakcji, prowadziła do jednego produktu - 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-2-okso-1,3,2-dioksafosfinanu 139b. Cykliczny fosfinan 139b, otrzymany jako jedyny produkt, świadczy jednoznacznie o tym, że wewnątrzcząsteczkowa cyklizacja zachodząca na etapie jodofosforanu (pierwszego produktu utlenienia) jest zdecydowanie dominującą reakcją następczą. W reakcji utleniania otrzymano analogiczne produkty wewnątrzcząsteczkowej cyklizacji, 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-2-okso-1,3,2-dioksafosfepan 139c i 2-[5'-O-(4,4'-dimetoksytrytylo)-tymidyn-3'-yloksy]-2-okso-1,3,2-dioksafosfokan 139d, odpowiednio dla pochodnych 4-hydroksybutylowej 142c i 5-hydroksypenylowej 142c, ale tylko w przypadku, gdy reakcje utleniania prowadzono w warunkach bezwodnych.
Ta sama reakcja przeprowadzona wobec dużego nadmiaru (30 ekw.) wody w przypadku diestru 142c prowadziła do fosfepanu 139c (~ 30%, 31P NMR) i 3-hydroksybutylofosforanu nukleozydu 141c (~ 70%). Stosowanie większych nadmiarów wody (60 ekw.) nie zmieniło proporcji ilościowych otrzymywanych produktów 139c i 141c. Utlenianie 5-hydroksypentylowej pochodnej 142d wobec 30 ekw. wody prowadziło do powstawania pożądanego 5-hydroksypentylofosforanodiestru nukleozydu 141d jako jedynego produktu reakcji.
Podsumowując, diestry fosforanowe typu 141 o potencjalnym zastosowaniu syntetycznym można otrzymywać z dobrymi wydajnościami utleniając wobec nadmiaru wody produkt kondensacji (wobec NEP-Cl) monoestru 121 z diolami wyższymi niż butanodiol. Ze względu na silną tendencję do cyklizacji nie można stosować dioli niższych (n < 5).
142b -
d
|
|
|
|
|
Schemat 26
