
| Czas reakcji [min.] |
|
|
|
|
| Stopień fosfonylacji [%] |
|
|
|
|
Badając przebieg reakcji w czasie (Tabela 6),
obserwowałem powstawanie nowego produktu oligonukleotydowego o mobilności
elektroforetycznej spodziewanej dla 5'-fosfonylowanego oligomeru II.
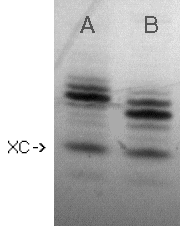
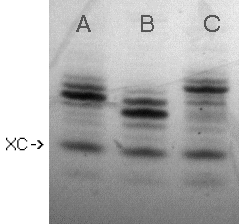
Po 120 min. substrat I (Zdjęcie 1,
linia A* ) ulegał praktycznie ilościowej fosfonylacji (Zdjęcie
1, linia B). Być może zastosowanie roztworu kwasu pirofosfonowego
o wyższych stężeniach przyspieszyłoby reakcję fosfonylacji, jednakże próby
przygotowania takiego roztworu zakończyły się niepowodzeniem, gdyż przy
stężeniach wyższych niż 0,2 M sól pirydyniowa kwasu pirofosfonowego wytrącała
się z roztworu.
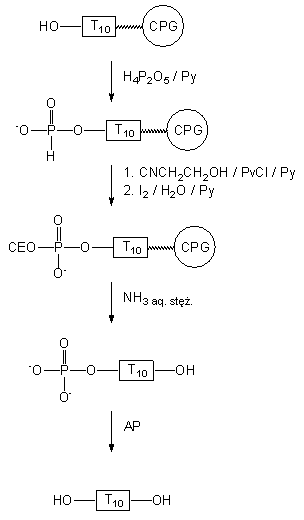
Chcąc uzyskać bardziej jednoznaczne potwierdzenie obecności reszty
5'-H-fosfonianowej w oligomerze II,
po odcięciu z podłoża i odblokowaniu (wodny stężony roztwór NH3)
poddałem go działaniu alkalicznej fosfatazy. 5'-Fosfonylowany T10
II
nie
ulegał defosfonylacji (PAGE), gdyż enzym nie rozpoznaje monoestrowej reszty
H-fosfonianowej jako substratu.87 Jeżeli 5'-fosfonylowany oligomer
II
w
reakcji kondesacji z 2-cyjanoetanolem (wobec Piv-Cl) przekształcono w diester
H-fosfonianowy, a po jego utlenieniu (I2 w wodnej pirydynie)
w fosforanodiester
III, który po odblokowaniu
(wodny stężony NH3) do IV
traktowano alkaliczną fosfatazą, to otrzymano ilościową defosforylację
IV
do nonafosforanu dekatymidynylowego
V
(Schemat 27). Powyższe wyniki można uznać
za wystarczający dowód na to, iż reakcja fosfonylacji oligomeru I
prowadzi do 5'-fosfonylowanego oligonukleotydu II.
 |
I II
III
IV
V |
Schemat 27
Tak więc, kwas pirofosfonowy może być z powodzeniem stosowany jako łagodny, monofunkcyjny czynnik fosfonylujący do wprowadzania reszt H-fosfonianowych na koniec 5' oligonukleotydów związanych z podłożem stałym.
Powstawanie dimerów można było zaobserwować podczas
opisywanej wcześniej fosfonylacji 5'-O-(4,4'-dimetoksytrytylo)tymidyny
w roztworze (str. *).
Ilość tego typu produktu zmniejszała się wraz ze wzrostem nadmiaru fosforynu
difenylowego użytego w reakcji. Można oczekiwać, że również w reakcjach
na podłożu stałym, stosując odpowiednie nadmiary czynnika fosfonylującego,
dimeryzacja będzie ograniczona lub wyeliminowana.
Tabela 7. Fosfonylacja T10 (I) fosforynem difenylowym
| Stężenie (PhO)2P(O)H [M] |
|
|
|
|
| Stopień fosfonylacji [%] |
|
|
|
|
| Stopień dimeryzacji [%] |
|
|
|
|
W pierwszej kolejności badałem możliwość powstawania dimerów T10-O-P(O)H-O-T10 w zależności od stosowanych nadmiarów fosfonianu difenylowego (Tabela 7). Oligomer I (Zdjęcie 2, linia A) poddałem działaniu roztworów fosforynu difenylowego w pirydynie o stężeniach 0,01, 0,05, 0,1 i 0,5M. Po 15 min. reakcji (czas wybrany arbitralnie), hydrolizie grupy fenoksylowej z powstałego 5'-fenylo-H-fosfonianu oligonukleotydu (Py-H2O 9:1, 30 min.) i jego odblokowaniu (wodny stężony roztwór NH3), produkty analizowano na żelu poliakrylamidowym. Pełna fosfonylacja następowała już przy użyciu 0,1M roztworu fosforynu, jednakże obok pożądanego H-fosfonianu oligonukleotydu obecne były znaczące (ok. 10%) ilości 5'-5' dimeru. Dopiero zastosowanie roztworu 0,5 molowego pozwoliło uzyskać H-fosfonian oligomeru (II) jako praktycznie jedyny produkt (Zdjęcie 2, linia B).

W drugiej kolejności badałem czas konieczny do całkowitej fosfonylacji
dla stężenia 0,5 M fosforynu difenylowgo, gdyż wybrany arbitralnie w pierwszym
doświadczeniu czas piętnastu minut mógł okazać się niepotrzebnie zbyt długi.
Stosując podobną metodę analizy (PAGE) mogłem ustalić, że przy powyższym
nadmiarze fosforynu difenylowego, reakcja fosfonylacji oligomeru T10
(I, Schemat 27) do 5'-H-fosfonianu
oligonukleotydu II jest zakończona
w czasie 5 minut (Tabela 8).
| Czas reakcji [min.] |
|
|
|
| Stopień fosfonylacji [%] |
|
|
|
Tak więc, fosforyn difenylowy okazał się czynnikiem pozwalającym na wydajną fosfonylację oligonukleotydów w czasie znacznie krótszym, niż w przypadku analogicznej reakcji z użyciem kwasu pirofosfonowego. Ponadto, fosforyn difenylowy jest tanim, handlowym odczynnikiem i można go używać bez dodatkowych przygotowań.
Pragnę zwrócić uwagę, że w wyniku reakcji fosfonylacji oligonukleotydu fosforynem difenylowym otrzymuje się 5'-fenylo-H-fosfonian oligonukleotydu, który zawiera podatną na transestryfikację grupę fenylową. Jeżeli zamiast hydrolizy tego produktu, podda się go reakcji z aminoalkoholem, można spodziewać się powstawania 5'-aminoalkilo-H-fosfonianu oligonukleotydu, będącego docelowym produktem tego etapu modyfikacji oligomeru. Byłby to kolejny, bardzo prosty, wariant funkcjonalizacji oligonukleotydów.
 |
|
|
 |
|
|
Jednakże po to, by zapewnić wysoką wydajność reakcji w układzie dwufazowym (roztwór/podłoże stałe), koniecznym jest stosowanie dużych nadmiarów (kilkadziesiąt, a nawet kilkaset ekwiwalentów molowych) reagentów. Stosowanie wolnego aminoalkoholu w tak dużym nadmiarze mogłoby powodować degradację otrzymanego metodą H-fosfonianową oligomeru (transestryfikacja internukleotydowych H-fosfonianodiestrów) lub powodować częściowe usuwanie ochronnych grup 2-cyjanoetylowych w oligonukleotydzie zsyntetyzowanym metodą amidofosforynową. Aby zapobiec tym niepożądanym reakcjom ubocznym, ponownie wykorzystałem protonowanie funkcji aminowej aminoalkoholu, które, jak będzie to później wykazane (przy reakcjach kondensacji oksydatywnej, str. *), znacznie osłabia nukleofilowość grupy -NH2 aminoalkoholu. Jednocześnie funkcja hydroksylowa zachowuje charakter nukleofilowy i w planowanej reakcji grupa ta powinna być nukleofilem dominującym.
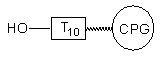
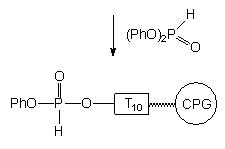
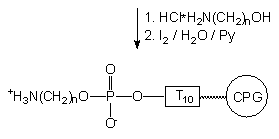
Po utlenieniu i odblokowaniu roztworem amoniaku, pożądany 5'-aminoalkilo-oligonukleotyd został otrzymany z wydajnością prawie ilościową (Zdjęcie 3, linia C). Podobne wyniki uzyskane zostały dla aminoalkoholi 153e i f. Jeśli jednak do takiej samej funkcjonalizacji wykorzystany został oligomer I uzyskany metodą amidofosforynową to, po odblokowaniu amoniakiem, obok właściwego produktu pojawiły się znaczące ilości (40 - 80%) oligomeru o innej mobilności elektroforetycznej. Produkt ten nie był natomiast obserwowany, kiedy do końcowego odblokowania zastosowana została metyloamina125(40% aq.) zamiast amoniaku. Problem tworzenia różnych produktów odblokowania 5'-aminoalkilofosforanów oligonukleotydów w zależności od metody ich syntezy omówię bardziej szczegółowo w dalszej części rozprawy.
 |
|
|
 |
|
|
 |
|
|
Schemat 29
Ze względów omówionych wcześniej, do transestryfikacji fenylowego H-fosfonianu oligonukleotydu VII stosowałem chlorowodorki aminoalkoholi. W pierwszej kolejności, stosując chlorowodorek aminopentanolu 153d (0,2M roztwór w pirydynie) badałem przebieg reakcji transestryfikacji VIIw czasie (Tabela 9). Po 60 minutach reakcja była zakończona, a jedynym nukleotydowym produktem obecnym w surowej mieszaninie po odcięciu z podłoża i odblokowaniu był sfunkcjonalizowany oligonukleotyd VI (PAGE).
Tabela 9. Transestryfikacja oligomeru VII chlorowodorkiem aminopentanolu
| Czas reakcji [min.] |
|
|
|
|
|
|
| Stopień funkcjonalizacji [%] |
|
|
|
|
|
|
Tak więc, w tym podejściu, nie stosując żadnych czynników kondensujących, a wykorzystując jedynie podatność arylowych H-fosfonianów na substytucję, można było w jednym ciągu reakcji przeprowadzić fosfonylację (I -> VII) i funkcjonalizację (VII -> VI) oligonukleotydu.
Podsumowując, do funkcjonalizacji oligonukleotydów powyższą metodą należy przeprowadzić następujące reakcje:
Metoda ta będąca odmianą Wariantu I, angażująca wyjątkowo
proste, dostępne handlowo reagenty pozwala na funkcjonalizację oligonukleotydów
w sposób jeszcze prostszy i szybszy od zaproponowanego w poprzednim
rozdziale niniejszej rozprawy.
Uważam, że obydwie opisane powyżej metody są równocenne i całkowicie spełniają początkowe wymogi funkcjonalizacji oligonukleotydów według Wariantu I. Fakt, że stosują one różnego typu substraty, a także różnego typu reakcje na poszczególnych etapach, może okazać się pomocny w przypadkach wykluczających stosowanie w trakcie funkcjonalizacji któregoś z reagentów (np. chlorofosforanów jako czynników kondensujących lub H-fosfonianu difenylowego wobec silnych zasad). Tak więc, w mojej ocenie, należy traktować powyższe metody jako uzupełniające, a nie alternatywne.
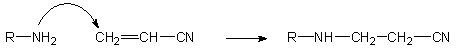
Schemat 30
Dla potwierdzenia tych założeń, otrzymany metodą H-fosfonianową i oczyszczony (PAGE) 5'-aminoalkilo-T10 traktowano wodnym roztworem amoniaku wobec nadmiaru akrylonitrylu (10 ekw.). Po 2 godz. reakcji 5'-aminoalkilo oligonukleotyd przekształcał się ilościowo w produkt o identycznej mobilności (PAGE), jak produkt uboczny, który powstawał w trakcie odblokowania oligomerów syntetyzowanych metodą amidofosforynową. Ten wynik silnie sugeruje, że wolna funkcja aminowa grupy 5'-aminoalkilowej sfunkcjonalizowanego oligomeru ulega N-alkilowaniu akrylonitrylem do analogu N-(2-cyjanoetylo)aminoalkilowego. Jeśli tak jest rzeczywiście, to zablokowanie funkcji aminowej grupą ochronną powinno całkowicie zapobiegać N-alkilowaniu. I faktycznie, oligomer z grupą 5'-[N-(4,4'-dimetoksytrytylo)]-aminoalkilową, traktowany takim samym roztworem amoniaku wobec nadmiaru akrylonitrylu, pozostał niezmieniony w ciągu 16 godzin.
Można przyjąć następne założenie, że zastosowanie do końcowego odblokowania oligomeru aminy (np. metyloaminy) w miejsce amoniaku powinno także zabezpieczyć przed N-alkilowaniem reszty aminoalkilowej, gdyż powstający akrylonitryl będzie wyłapywany przez znajdującą się w dużym nadmiarze aminę. Także i wtym przypadku założenia zweryfikowano w doświadczeniu, w którym oczyszczony 5'-aminoalkilo-oligonukleotyd pozostał niezmieniony podczas szesnastogodzinnego działania akrylonitrylu (10 ekw.) w wodnym 40% roztworze metyloaminy.
Aby uzyskać bezpośrednie dowody możliwości zachodzenia reakcji alkilowania grup aminowych, przeprowadziłem badania na związkach modelowych: fosforanie nukleozydowo - aminoalkilowym 146 i aminoalkilonukleozydzie 14899 (Schemat 31).
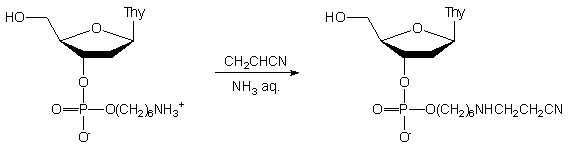
|
|
|
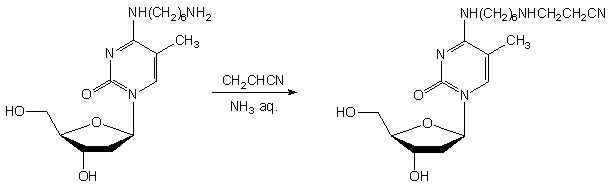
|
|
|
Schemat 31
Oba te związki traktowane akrylonitrylem (10 ekw.) w roztworze amoniaku, w ciągu 15 min. przekształcały się ilościowo w nowe produkty (TLC). Po izolacji, ustalono ich strukturę (1H i 31P NMR) jako analogi N-(2-cyjanoetylo)aminoalkilowe 147 i 149. W podobnych doświadczeniach z zastosowaniem metyloaminy wmiejsce amoniaku, oba związki (146 i 148) pozostały niezmienione w ciągu 24h.
Podsumowując, podczas otrzymywania sfunkcjonalizowanych oligomerów otrzymanych metodą amidofosforynową, zawierających wolne reszty aminoalkilowe, należy mieć na uwadze możliwość alkilacji tych reszt akrylonitrylem, uwolnionym w trakcie końcowego odblokowania amoniakiem. Reakcja ta nie zachodzi gdy:
