
Jednym z podstawowych warunków efektywnego sprzęgania oksydatywnego jest bezwodność środowiska reakcji. Śladowe ilości wody z substratów tj. diestru 150 i aminoalkoholu 124e usuwałem przez odparowanie z nadmiarem bezwodnej pirydyny. Okazało się, że w trakcie takiej procedury przygotowywania substratów następuje transestryfikacja (!) z uwolnieniem nukleozydu i nienukleotydowych produktów zawierających fosfor. Na ten łatwo zachodzący, niepożądany proces zwróciłem szczególną uwagę, gdyż sądzę, że może on mieć bardziej ogólne konsekwencje w chemii H-fosfonianów nukleozydów czy diestrów H-fosfonianowych w ogóle. Dlatego też podjąłem bardziej szczegółowe badania nad transestryfikacją H-fosfonianów alkoholami, alkoholami wobec amin oraz aminoalkoholami, a ich wyniki opisałem w dalszej części rozprawy.
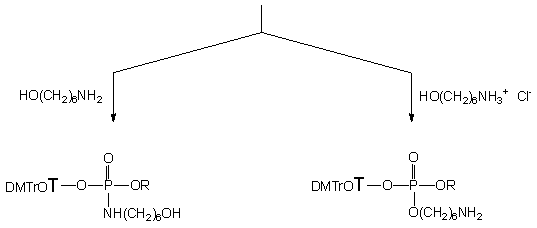
Przygotowując substraty 150 i 124e osobno można zapobiec transestryfikacji i dalej prowadzić reakcję oksydatywnej kondensacji (Schemat 32). Diester 150 poddany utlenianiu jodem (1 ekw.) w pirydynie w obecności 6-aminoheksan-1-olu 124e (10 ekw.) w czasie ~ 15 s tworzy jeden produkt o przesunięciu chemicznym ~ 10 ppm (31P NMR). Związek ten, po izolacji zidentyfikowałem jako 6-hydroksyheksyloamidofosforan nukleozydowo - etylowy 154 (31P i 1H NMR, TLC i analiza elementarna). Obecność amidofosforanu 154 jako jedynego produktu wskazuje, że w warunkach reakcji funkcja aminowa jest zdecydowanie efektywniejszym nukleofilem wobec powstałego in situ jodofosforanu niż funkcja hydroksylowa aminoalkoholu. Dla realizacji celów tej pracy korzystniejsze byłoby odwrócenie chemoselektywności omawianej reakcji i otrzymywanie triestrów aminoalkilowych. Jest to możliwe np. przez zablokowanie funkcji aminowej aminoalkoholu, co wiąże się jednak z dodatkowym przygotowaniem aminoalkoholu, a także koniecznością selektywnego odsłonięcia funkcji aminowej. Można także zakładać prostsze rozwiązanie, w którym nukleofilowość atomu azotu funkcji -NH2 będzie znacznie zredukowana przez jej protonowanie, podczas gdy charakter nukleofilowy reszty hydroksylowej aminoalkoholu pozostaje praktycznie niezmieniony.
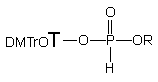
150 151 152
|
124e |
 |
153e
|
|
154 155156
|
157 158 159 |
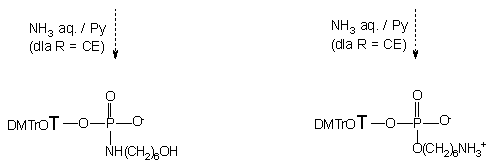
|
160
|
132e |
| 150,
154,
157,R
= etyl
151, 155, 158, R = 3'-O-(4,4'-dimetoksytrytylo)tymidyn-5'-yl 152, 156, 159,R = 2-cyjanoetyl |
Schemat 32
Schemat 32
Założenie to zweryfikowałem pozytywnie w reakcji diestru 150 z chlorowodorkiem aminoheksanolu 153e w warunkach utleniania identycznych jak w przypadku aminoalkoholu nieprotonowanego. Reakcja zachodziła zdecydowanie wolniej (30 min.) i prowadziła do jednego produktu o przesunięciu chemicznym dP >>-2 ppm (typowym dla fosforotriestrów), który wyizolowano i określono jego strukturę (31P i 1H NMR, TLC i analiza elementarna) jako etylo-6-aminoheksylo-3'-fosforan nukleozydu 157. Tak więc, proste protonowanie funkcji -NH2 aminoalkoholu rzeczywiście odwraca jego reaktywność na korzyść grupy -OH i pozwala ukierunkować reakcję oksydatywnej kondensacji na tworzenie fosforotriestrów jako jedynych produktów reakcji. Zgodnie z moją najlepszą wiedzą, jest to pierwszy przykład otrzymywania fosforanotriestrów aminoalkilowych na drodze utleniania diestrów H-fosfonianów wobec nieblokowanych (a jedynie N-protonowanych) aminoalkoholi.
Biorąc pod uwagę brak odpowiedniej metody selektywnego usuwania reszty etylowej oraz nietrwałość fosforotriestrów w standardowych warunkach odblokowania oligonukleotydów (stężony wodny amoniak), syntetyczna użyteczność etylowych triestrów typu 157 jest ograniczona. Dlatego też podjąłem badania nad oksydatywną kondensacją aminoalkoholi z H-fosfonianem dinukleozydowym typu 151, a także z 2-cyjanoetylo-H-fosfonianem nukleozydu typu 152. Dinukleozydo-H-fosfonian 151 utleniany jodem wobec 6-aminoheksan-1-olu i jego chlorowodorku prowadził, w porównywalnym czasie, do analogicznych produktów oksydatywnej kondensacji jak pochodna etylowa 150 tj. 6-hydroksyheksyloamidofosforanu 155 i 6-aminoheksylofosforo triestru 158. W przypadku diestru 152 niosącego b -eliminacyjną grupę 2-cyjanoetylową, jeżeli reakcję utleniania wobec wolnego aminoheksanolu 124e prowadzono w pirydynie to obok spodziewanego produktu kondensacji 156 znajdowano H-fosfonian nukleozydu 121 (~ 20%, 31P NMR). Ten ostatni związek jest produktem reakcji b -eliminacji reszty 2-cyjanoetylowej z substratu 152 przed jego utlenieniem. Tak więc sądzić można, że w warunkach reakcji, b -eliminacja reszty 2-cyjanoetylowej z substratu 152 jest dostatecznie szybka by konkurować z utlenianiem, co znajduje odzwierciedlenie w kompozycji produktów (obecność H-fosfonianiu nukleozydu) po zakończeniu procesu utleniania. Ponadto, w mieszaninie poreakcyjnej widoczne były znaczące ilości (~ 20%) amidofosforanów nienukleotydowych, które były produktami oksydatywnej kondensacji dialkilo-H-fosfonianodiestrów, powstałych w szybkiej, poprzedzającej utlenienie, reakcji transestryfikacji związku 152 aminoalkoholem 124e.
Jeżeli tę samą reakcję utleniania 2-cyjanetylo-H-fosfonianu nukleozydu 152 wobec 124e przeprowadzano w acetonitrylu, to praktycznie jedynym (> 98%, 31P NMR) powstałym produktem był amidofosforan nukleozydu 156. Tak więc można wnioskować, że zmiana środowiska reakcji z zasadowego (pirydyna) na obojętne (acetonitryl) musiała znacznie spowolnić konkurencyjne reakcje transestryfikacji oraz b -eliminacji, przy zachowaniu szybkiego procesu utleniania i równie szybkiej następczej reakcji aminolizy powstałego jodofosforanu do amidofosforanu 156.
Zdecydowanie różne wyniki otrzymałem w reakcji utleniania 2-cyjanetylo-H-fosfonianu nukleozydu 152 wobec chlorowodorku 6-aminoheksanolu (153e). Reakcję prowadziłem w pirydynie ze względu na niedostateczną rozpuszczalność 153e w acetonitrylu. Pomimo zastosowania pirydyny jako medium reakcyjnego, protonowanie funkcji aminowej aminoalkoholu istotnie lub całkowicie zahamowało reakcje transestryfikacji czy b-eliminacji grupy 2-cyjanoetylowej. Reakcja jednak nie przebiegała czysto i oprócz aminoalkilofosforotriestru nukleozydu 159 (diastereoizomery, dP = -2,31 i -2,41 ppm) w mieszaninie reakcyjnej obecny był szereg produktów w zakresie -1 do -2 ppm. Całkowita transformacja substratu 152 zachodziła w czasie 30 minut. Prowadzenie reakcji wobec N-metyloimidazolu lub zmiana środowiska reakcji na 50% lutydynę w chlorku metylenu nie poprawiła znacząco obrazu reakcji. Natomiast w obecności stosunkowo niewielkich nadmiarów tetrazolu (4 ekw.) oksydatywne sprzęganie 152 i 153e prowadziło wyłącznie do triestru 159 i było zakończone w ciągu 5 minut. Jest bardzo prawdopodobne, że tetrazol działa tu jako nukleofil, przekształcając aktywne produkty utlenienia (jodofosforan czy addukt pirydyniowy fosforodiestru) w tetrazolid, który czysto i szybko reaguje z funkcją hydroksylową protonowanego aminoalkoholu.
Produkty 154, 155, 157 i 158 zostały oczyszczone chromatograficznie jako takie, natomiast pochodne 2-cyjanoetylowe 156 i 159 były niedostatecznie trwałe (b-eliminacja grupy 2-cyjanoetylowej) w trakcie izolacji. Dlatego też ze związków 156 i 159 usunięto grupę 2-cyjanoetylową (NH3 aq. stęż./pirydyna), po czym izolowano trwałe produkty końcowe: 6-hydroksyheksyloamidofosforan nukleozydu 160 i 6-aminoheksylofosforan nukleozydu 132e.
Powyższe prace nad oksydatywną kondensacją H-fosfonianowych diestrów nukleozydów z aminoalkoholami są przykładem kontrolowania kierunku reakcji przez zmianę nukleofilowości funkcji aminowej aminoalkoholu na drodze prostego protonowania bez konieczności jej dodatkowego blokowania. Otwiera to możliwości stosowania jednego typu reakcji do efektywnej syntezy analogów nukleotydów niosących reszty hydrokyalkiloamidofosforanowe lub aminoalkilofosforanowe. Wyniki tych prac zostały opublikowane w 1995 roku.146
W trakcie realizacji tej części rozprawy moją szczególną uwagę zwróciły nieoczekiwanie łatwo i spontanicznie zachodzące reakcje transestryfikacji H-fosfonianodiestrów aminoalkoholami. W dostępnej mi literaturze znalazłem bardzo nieliczne prace128-131 dotyczące tego typu reakcji. Jak już wcześniej wspomniałem, sądzę, że reakcje te mogą mieć bardziej ogólne konsekwencje w chemii nukleotydów i oligonukleotydów, dlatego też poświęciłem im następny rozdział rozprawy.
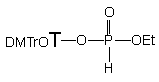
150
|
|
|
|
|
163
|
|
|
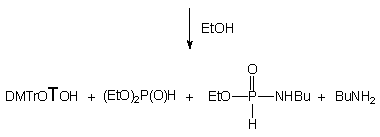
|
119
|
|
|
Schemat 33
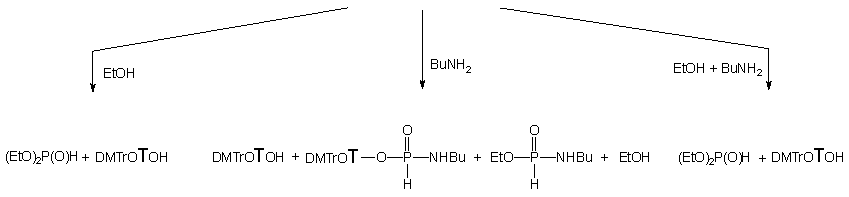
Jednym z istotnych czynników warunkujących szybkość omawianej reakcji może być zasadowość środowiska, wpływająca zarówno na aktywację alkoholu (generowanie jonów alkoksylowych) jak i na ternalizację substratu 150 do tri-skoordynowanego dialkilofosforynu, który powinien być bardziej podatny na substytucję na centrum fosforowym niż ten sam związek w formie H-fosfonianu. Przypuszczenie to potwierdziłem w reakcji diestru 150 z etanolem wobec trietyloaminy (10 ekw.), w których to warunkach reakcja transestryfikacji do fosfonianu dietylowego była zakończona po 60 godzinach.
Wyniki powyższych doświadczeń wskazują na to, iż transestryfikacje H-fosfonianodiestrów alkoholami w umiarkowanie zasadowych warunkach zachodzą bardzo wolno, i że można je przyspieszać wprowadzając do reakcji mocne zasady.
Podsumowując, reakcje aminolizy H-fosfonianu nukleozydowo - etylowego 150 zachodzą szybciej niż analogiczne reakcje transestryfikacji etanolem. Jednakże chciałbym zwrócić szczególną uwagę na fakt, że uwolniony w trakcie aminolizy alkohol jest na tyle silnie aktywowany przez aminę, że w mieszaninie reakcyjnej obserwuje się produkty transestryfikacji z jego udziałem jako główne, trwałe produkty finalne.
Reakcja H-fosfonianu nukleozydowo - etylowego 150 z ekwimolarną ilością etanolu i butyloaminy (po 10 ekw.) zachodziła kilkakrotnie szybciej niż te, w których etanol i amina były stosowane oddzielnie. W czasie dziesięciu godzin substrat przereagował całkowicie, a jedynymi otrzymanymi produktami był fosfonian dietylowy 161 i nukleozyd 119 (Schemat 33). Monitorując (31P NMR) reakcję w czasie nie stwierdziłem powstawania teoretycznie możliwych pośrednich produktów aminolizy. Dodatkowym potwierdzeniem braku udziału amidofosfonianu nukleozydu 163 jako produktu pośredniego jest wcześniej opisana jego reakcja w podobnym układzie, w której powstają trwałe produkty - H-fosfonian dietylowy 161 i amidofosfonian etylowy 163. Ponieważ w omawianym doświadczeniu tego ostatniego związku nie obserwowano, pozwala to twierdzić, że etanol silnie aktywowany butyloaminą (generowanie jonu etoksylowego) jest reagentem zdecydowanie dominującym, w wyniku czego otrzymuje się bezpośrednio produkt transestryfikacji 161 bez udziału amidofosfonianów nukleozydów typu 163 jako produktów pośrednich.
W przypadku aminoetanolu 124a po
2 godz. stwierdziłem całkowite przereagowanie substratu 150
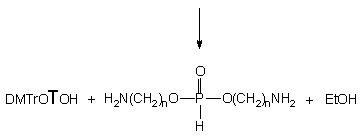
do nienukleotydowego di(aminoetylo)-H-fosfonianu 165a i nukleozydu
119
(Schemat 34). Śledzenie przebiegu reakcji (31P NMR) w czasie
ujawniło powstawanie jednego produktu pośredniego - aminoetylo-H-fosfonianu
etylowego 164a. Wydawać się więc mogło, że reakcja zachodzi
chemoselektywnie z podstawieniem nukleozydu. Jednakże eksperyment, w którym
aminoetylo-H-fosfonian nukleozydu 134a (otrzymany przez transestryfikację
arylo-H-fosfonianu nukleozydowego; omówienie w dalszej części rozprawy)
traktowano nadmiarem aminoetanolu (10 ekw.), wykazał że pochodna 134a
ulega transestryfikacji do symetrycznego diestru 165a w czasie
2 min. Jeżeli w szeregu reakcji 150 -> 134a
-> 165a pierwsza reakcja (150 -> 134a)
jest wolna, a reakcja następcza (134a -> 165a)
zdecydowanie szybsza, to nie będziemy obserwować pośredniego produktu 134a,
gdyż z chwilą jego powstania będzie on ulegał dalszej transestryfikacji
do finalnego produktu 165a. Nie można więc wykluczyć, że
omawiana reakcja 150 -> 165a zachodzi dwukierunkowo,
z udziałem diestrów 134a i 164a jako produktów
pośrednich, przy czym w trakcie reakcji obserwujemy jedynie ten ostatni
związek, gdyż wolniej ulega on dalszej transestryfikacji do symetrycznego
diestru 165a.
150
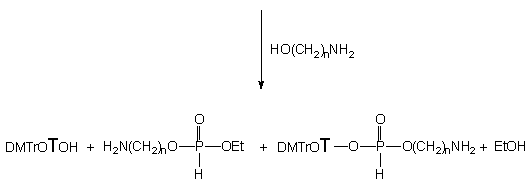
|
|
|
134
|
|
|
|
Schemat 34
Wyższe homologi aminoetanolu, 3-aminopropan-1-ol (124b), 4-aminobutan-1-ol (124c) i 6-aminoheksan-1-ol (124e) reagowały podobnie, jednak w trakcie reakcji jako produkty pośrednie, obok H-fosfonianów typu 164, widoczne były diestry nukleozydowo - aminoalkilowe typu 134. W miarę wydłużania się łańcucha polimetylenowego szybkości reakcji stopniowo malały, zbliżając się do szybkości reakcji mieszaniny etanolu i n-butyloaminy. Czasy całkowitego przereagowania substratu 150 wynosiły 3 h dla 124b, 4 h dla 124c i 5 h dla 124e. Przebieg poszczególnych reakcji w czasie przedstawiono na Wykresie 1.
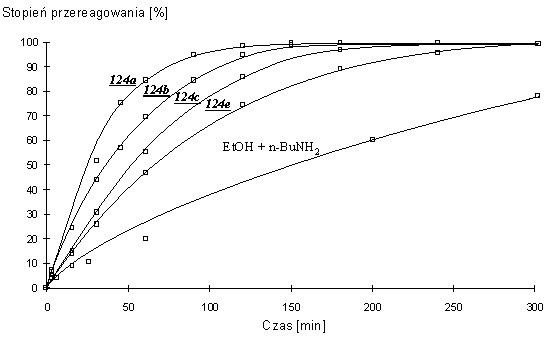
Wykres 1. Czas rozkładu H-fosfonianu nukleozydowo - etylowego 150 wobec aminoalkoholi (10 ekw.) w pirydynie
Produktami końcowymi transestryfikacji były symetryczne diestry typu 165 i nukleozyd 119. W przypadku pochodnych aminoetylowych i aminobutylowych diestry typu 134, 164 i 165 ulegały powolnej przemianie do produktów rezonujących w zakresie 3 - 4 ppm, których struktury nie udało mi się ustalić.
Otrzymane wyniki wskazują, że reaktywność aminoalkoholi maleje wraz ze wzrostem długości łańcucha polimetylenowego oddzielającego funkcje hydroksylową od aminowej i jest odwrotnie proporcjonalna do ich zasadowości (Tabela 10). Ponadto, podobnie jak w doświadczeniach z mieszaną alkoholu i aminy, w żadnym z przypadków nie znaleziono produktów zawierających wiązania P-N.
Tabela 10. Wartości pKa133 i czasy reakcji z diestrem 150 wybranych aminoalkoholi i etanolu wobec amin
|
|
|
|
| aminoetanol 124a | 9,51 |
|
| aminopropanol 124b | 9,96 |
|
| aminobutanol 124c | 10,35 |
|
| aminopentanol 124d | 10,46 |
|
| aminoheksanol 124e | 10,60 |
|
| n-butyloamina | 10,66 |
|
| trietyloamina | 11,01 |
|
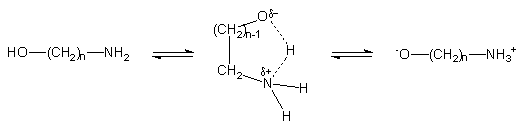
Taki stan rzeczy można tłumaczyć udziałem formy jonu obojnaczego w strukturze aminoalkoholi (Schemat 35). Zakładając, że efektywnym nukleofilem w reakcji transestryfikacji jest jon alkoksylowy aminoalkoholu, to szybkość reakcji transestryfikacji będzie wzrastała wraz ze wzrostem udziału formy zwitterjonu 167 w strukturze aminoalkoholi. Można sądzić, że udział tej formy będzie wzrastał wraz z zasadowością grupy -NH2 aminoalkoholu. Jednakże eksperymenty wykazały, że modelowy diester 150 ulega transestryfikacji najszybciej w przypadku aminoalkoholi o najniższej zasadowości tj. 124a i 124b. Jest bardzo prawdopodobne, że transfer protonu z funkcji hydroksylowej na aminową w aminoetanolu (124a) i 3-aminopropan-olu-1 (124b) zachodzi wewnątrzcząsteczkowo poprzez przejściowe 5-cio- (dla 124a) i 6-cio- (dla 124b) członowe struktury cykliczne typu 166, co może znacznie ułatwiać przechodzenie w formę jonów obojnaczych typu 167. W przypadkach, gdzie funkcje aminową i hydroksylową oddziela większa liczba grup metylenowych, udział wewnątrzcząsteczkowych struktur cyklicznych zanika i przechodzenie w formę jonu obojnaczego odbywa się poprzez mniej efektywne oddziaływania międzycząsteczkowe. W rezultacie, pomimo wyższej zasadowości aminoalkoholi 124c i 124e, stężenie postulowanego zwitterjonu jest niższe i w konsekwencji aminoalkohole te są mniej reaktywne w reakcjach transestryfikacji. Postulat jonu alkoksylowego jako najbardziej reaktywnego nukleofila może tłumaczyć powstawanie w reakcjach transestryfikacji wyłącznie fosforestrów, a proponowane różnice trwałości takich struktur tłumaczyłyby szereg reaktywności aminoalkoholi.
|
|
|
|
Schemat 35
Jednoznaczne potwierdzenie występowania struktur jonów podwójnych w aminoalkoholach byłoby, jak sądzę, bardzo trudne. Chcąc chociaż częściowo dowieść poprawności powyższych założeń, wybrałem metodę pośrednią. Założyłem, że jeżeli stężenie jonu alkoksylowego aminoalkoholu odpowiedzialne jest za szybkość transestryfikacji, to wprowadzenie do środowiska reakcji mocnej zasady (trietyloaminy) powinno w przypadku aminoetanolu reakcję spowolnić (hamowanie tworzenia przejściowej struktury 166), a w przypadku aminoheksanolu przyspieszyć (trietyloamina jako silniejsza zasada niż grupa -NH2 aminoheksanolu może efektywniej deprotonować funkcję -OH). Wyniki takich doświadczeń, w których obok aminoalkoholi użyłem 10 ekw. trietyloaminy były nadspodziewanie zgodne z założeniami, gdyż szybkość reakcji transestryfikacji diestru 150 aminoetanolem zmniejszyła się (całkowite przereagowanie substratu - 3 godz.) a dla aminoheksanolu wzrosła (całkowite przereagowanie substratu - 3,5 godz.). Tak więc, trietyloamina znacznie osłabiła efekty wynikające ze struktury aminoalkoholi i w konsekwencji szybkości reakcji były porównywalne.
W przypadku aminoetanolu i w mniejszym stopniu aminopropanolu należy przedyskutować także sprawę wpływu efektu indukcyjnego na nukleofilowość atomu tlenu w bardzo różnych elektronowo tautomerach hydroksy-amino typu 124 i okso-amonio typu167. W tautomerze 124a i 124b funkcja -NH2 jest grupą elektrono-donorową wzmagającą nukleofilowość atomu tlenu reszty hydroksylowej. Natomiast protonowana funkcja aminowa w tautomerach 167a i 167b jest grupą elektrono-akceptorową. Efekt indukcyjny w przypadku struktur zwitterjonów 167a i 167b powinien obniżać ich reaktywność w porównaniu z anionami aminoalkoksylowymi [-O(CH2)nNH2]. Prawdopodobnie tak jest, ale ten sam efekt indukcyjny powinien także mieć wpływ stabilizujący tautomer okso-amonio typu 167, prowadząc do wzrostu jego globalnego stężenia w mieszaninie reakcyjnej, co w przypadku aminoalkoholi może być czynnikiem decydującym o szybkości transestryfikacji H-fosfonianodiestrów.
Mając powyższe na uwadze, sądzę, że różnice reaktywności różnych aminoalkoholi w reakcjach transestryfikacji są wypadkową powyżej dyskutowanych efektów i wynikają one z ich właściwości strukturalnych i fizykochemicznych.
Jedynym produktem otrzymanym w reakcji diestru 152 z nadmiarem (10 ekw.) n-butyloaminy był H-fosfonian nukleozydu 121, produkt b -eliminacji grupy 2-cyjanoetylowej. Całkowite przereagowanie substratu 152 obserwowano po 30 min., a w mieszaninie poreakcyjnej nie znaleziono (31P NMR) potencjalnie możliwych produktów aminolizy. Tak więc, reakcja b -eliminacji grupy 2-cyjanoetylowej butyloaminą z substratu 152 jest ok. dwustukrotnie szybsza niż prowadzona także w pirydynie reakcja transestryfikacji tego związku etanolem.
152
|
150
|
168
|
|
|
|
|
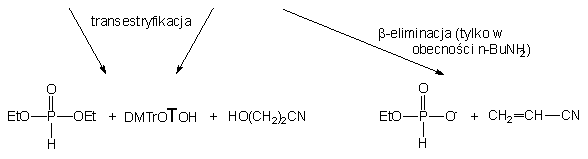
161 119 169 171 170
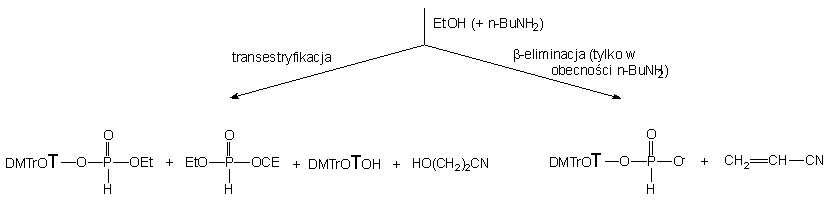
Biorąc pod uwagę powyższe wyniki, dość nieoczekiwane rezultaty otrzymałem, traktując diester 152 mieszaniną etanolu i butyloaminy (po 10 ekw.). W tej szybkiej reakcji (całkowite przereagowanie substratu po ~ 2 min.) powstały głównie produkty transestryfikacji 150 i 168 (90%), ponownie w stosunku 3 : 1 i jedynie niewielka ilość (10%) H-fosfonianu nukleozydu 121 (produkt b-eliminacji). Jak można było się spodziewać, produkty 150 i 168 ulegały następczym reakcjom do H-fosfonianu dietylowego 161 (transestryfikacja) i H-fosfonianu etylowego 171 (b-eliminacja). Podobnie jak w przypadku etylo-H-fosfonianu nukleozydu 150 traktowanym mieszaniną etanolu i butyloaminy, także i w tym eksperymencie wyraźnie zaznacza się przewaga reakcji transestryfikacji nad pozostałymi typami możliwych reakcji (aminoliza czy b -eliminacja).
Proponowana wcześniej interpretacja właściwości estryfikujących mieszanin alkoholi i amin potwierdziła się także i w przypadku 2-cyjanoetylowego diestru 152. Bardzo wyraźnie widoczne różnice szybkości reakcji transestryfikacji w porównaniu z analogiem etylowym 150 (2 min. vs 10 h) można przypisać oddziaływaniu reszty 2-cyjanoetylowej, która wzmaga elektrofilowość, a tym samym reaktywność centrum fosforowego substratu 152 w reakcjach substytucji nukleofilowej.
152
|
|
172
|
119
|
|
|
|
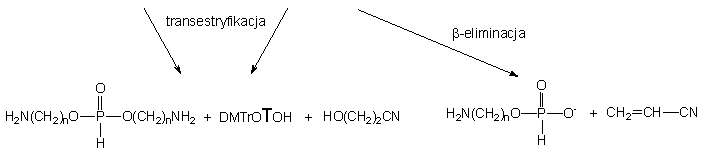
165 119 169 173 170 Schemat 37
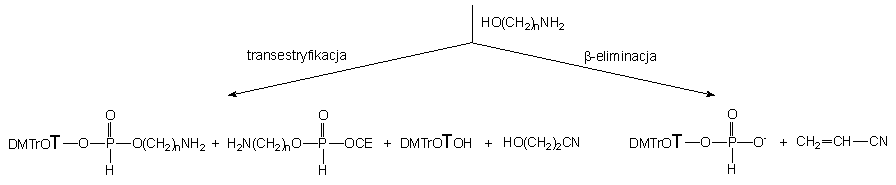
W przypadku reakcji 152 z aminoetanolem
w pierwszym zarejestrowanym widmie znaleziono cztery produkty - H-fosfonian
2-aminoetylowo - nukleozydowy 134a (25%), H-fosfonian 2-aminoetylowo
- 2-cyjanoetylowy 172a (5%) i produkt główny, symetryczny
H-fosfonian di(2-aminoetylowy) 165a (59%) oraz H-fosfonian
nukleozydu
121 (10%) (Schemat 37). H-Fosfonian nukleozydu
121
jest produktem reakcji b-eliminacji grupy
2-cyjanoetylowej z substratu 152, a diestry 134a,
172a
i 165a są produktami transestryfikacji. Na podstawie stosunków
ilościowych produktów poszczególnych typów reakcji można wnioskować, że
podobnie jak w przypadku stosowania mieszaniny etanolu i butyloaminy, reakcją
główną jest transestryfikacja (90%). Z kolei, stosunek ilościowy produktów
transestryfikacji 134a i 172a (5 : 1) wskazuje,
iż reszta 2-cyjanoetoksylowa łatwiej ulega substytucji niż nukleozyd. Przy
stosowanym nadmiarze 124a zachodzi szybka następcza reakcja
transestryfikacji diestrów 134a i 172a do symetrycznego
H-fosfonianu di(aminoalkilowego) 165a i równoległa, wolniejsza
reakcja b-eliminacji reszty 2-cyjanoetylowej
z 172a do monoestru 173a. Po ok. 10 min. finalnymi
produktami reakcji diestru 152 z 2-aminoetanolem były H-fosfonian
di(2-aminoetylowy) 165a oraz monoestry 121
i 173a.
W przypadku aminoalkoholi 124b, 124c i 124e obserwowano podobny przebieg transestryfikacji i równoległej b-eliminacji. Jednakże następcze reakcje transestryfikacji związków typu 134 i 172 do symetrycznych di(aminoalkilo)-H-fosfonianów typu 165 jako finalnych produktów zachodziły zdecydowanie wolniej (124b - 90 min., 124c - 120 min. i 124e - 180 min.) niż w przypadku 2-aminoetanolu.
151
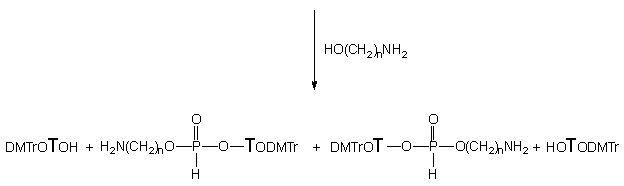
|
|
174 |
134
|
|
|
|
|
|
Schemat 38
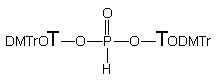
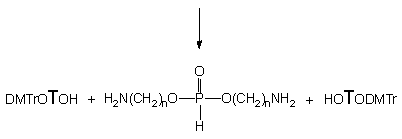
H-Fosfonian 151 traktowany aminoalkoholami 124a - c i 124e we wszystkich przypadkach ulegał na pierwszym etapie transestryfikacji do 3' i 5' aminoalkilo-H-fosfonianów nukleozydów typu 134 i 174 oraz nukleozydów 119 i 175. Całkowite przereagowanie substratu 151 przy dziesięciokrotnym nadmiarze molowym aminoalkoholu, wymagało dla 124a - 15 min., dla 124b - 60 min., dla 124c - 240 min. i dla 124e - 360 min. Z chwilą przereagowania substratu 151, głównymi produktami obecnymi w mieszaninach reakcyjnych były symetryczne diaminoalkilo-H-fosfoniany typu 165, produkty następczej reakcji transestryfikacji nukleotydowych pochodnych typu 134 i 174 powstałych w pierwszym etapie z substratu 151. Na uwagę zasługuje fakt, że produkty 134 i 174 powstawały w prawie równych ilościach, co świadczy o braku chemoselektywności transestryfikacji H-fosfonianu dinukleozydowego.
Strukturę nieznanych dotąd 5'-(aminoalkilo)-H-fosfonianów nukleozydu typu 174 ustaliłem przez porównanie z identycznymi produktami otrzymanymi inną metodą.134 Ze względu na ich nietrwałość, przed izolacją utleniałem je do trwałych aminoalkilofosforanów nukleozydów 176.
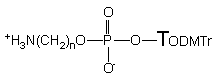
176
Dla sprawdzenia poprawności tych założeń, badałem
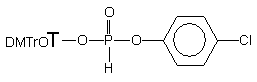
podatność p-chlorofenylo-H-fosfonianu nukleozydu 177
na substytucję alkoholami, aminami i aminoalkoholami (Schemat 39). Ze względu
na nietrwałość diestru 177 w warunkach przerobu i izolacji,
wygenerowałem go in situ przez kondensację p-chlorofenolu
i H-fosfonianu nukleozydu 121 wobec chlorku piwaloilu w pirydynie
(kondensacja standardowa).
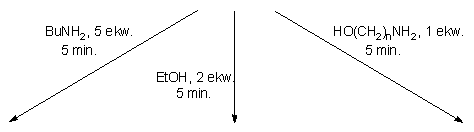
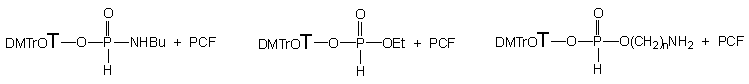
|
|
|
|
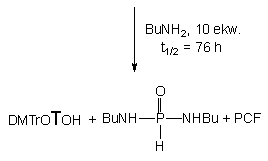
119 178
W przypadku etanolu wystarczył niewielki jego nadmiar (2 ekw.) by transestryfikacja diestru arylowego 177 zaszła ilościowo w ciągu zaledwie 5 minut. Reakcja przebiegała czysto i z pełną chemoselektywnością do H-fosfonianu etylowo - nukleozydowego 150.
Aminoalkohole 124a - c i e, bez względu na długość mostka polimetylenowego i używane w ilościach ekwimolarnych, reagowały z substratem 177 równie szybko (5 min.), tworząc wyłącznie produkty transestryfikacji - odpowiednie aminoalkilo-H-fosfoniany nukleozydów 134a - c i e. Całkowitą chemoselektywność tej reakcji można tłumaczyć udziałem postulowanej wcześniej formy zwitterjonu aminoalkoholu. Jednakże, należy rozważyć także drugą możliwość tj. fakt, że aminoalkohol dodawany był do mieszany reakcyjnej, w której po kondesacji fenolu i H-fosfonianu nukleozydu 121, obecne były kwasowe produkty (chlorowodorek pirydyny, kwas piwalowy). W takich warunkach aminoalkohol mógł ulegać szybkiemu N-protonowaniu (osłabienie nukleofilowości funkcji -NH2) i tej formie reagować z diestrem 177 wyłącznie do produktu estrowego 134, a nie amidofosfonianu. Bez względu na to, który z czynników decyduje o chemoselektywności omawianej reakcji, jej produktami są aminoalkilo-H-fosfoniany nukleozydów typu 134 i powyższa procedura może być uznana jako alternatywna (vs opisana w poprzedniej części rozprawy, bezpośrednia kondensacja aminoalkoholi i H-fosfonianów nukleozydów aktywowanych chlorofosforanami), bardzo efektywna metoda syntezy związków typu 134 i ich pochodnych.
Arylo-H-fosfonian nukleozydu 177 traktowany n-butyloaminą (5 ekw.) reagował tak samo szybko (5 min.) jak etanol czy aminoalkohole i także z całkowitą chemoselektywnością, tworząc jeden produkt - amidofosfonian nukleozydu 162. Zakładając, że większa część (~ 3 ekw.) użytego nadmiaru aminy w wyniku protonacji (obecne po kondensacji, chlorowodorek pirydyny i kwas piwalowy) mogła być przekształcona w mniej aktywną sól amoniową, to pozostały niewielki nadmiar wolnej aminy (~ 2 ekw.) wystarczył do szybkiej aminolizy substratu 177. Mając na uwadze szybkość, chemoselektywność i czystość omawianej reakcji można polecać arylo-H-fosfoniany nukleozydów jako uniwersalne substraty do syntezy amidofosfonianów nukleozydów typu 162, które z nielicznymi wyjątkami135 były jak dotąd niedostępne.
Stosowanie większych nadmiarów etanolu i aminoalkoholi powodowało wcześniej już opisane reakcje następcze prowadzące do nienukleotydowych, symetrycznych diestrów 161 i 165. Przy dużych nadmiarach (10 - 20 ekw.) butyloaminy, powstały na pierwszym etapie amidofosfonian nukleozydu 162 ulegał dalszej powolnej (t1/2 = 76 h) aminolizie do symetrycznego diamidofosfonianu 178.
Właściwości p-chlorofenylo-H-fosfonianu nukleozydu 177 w reakcjach transestryfikacji i aminolizy w pełni potwierdziły założenia i spodziewane efekty związane z wpływem podstawnika przy atomie fosforu. Reakcje te zachodziły szybko i całkowicie chemoselektywnie, co należy przypisać zarówno aktywacji centrum fosforowego przez wyciągającą elektrony grupę p-chlorofenylową, jak i jej właściwościom jako grupy opuszczającej w reakcjach substytucji.
Z przedstawionych powyżej wyników badań nad reakcjami transestryfikacji i aminolizy diestrów H-fosfonianów wysnuć można kilka wniosków. Uważam, że spontaniczność tych reakcji wynika z charakteru wiązania H-fosfonianodiestrowego. Manewrując środowiskiem lub podstawnikami przy centrum fosforowym można przebieg reakcji kontrolować i ukierunkować. Umożliwia to racjonalne projektowanie substratów (np. arylo-H-fosfonianów nukleozydów), które w łagodnych warunkach pozwalają otrzymywać pochodne trudno osiągalne innymi metodami. Ponadto, otrzymane rezultaty pozwoliły mi dostrzec niebezpieczeństwa i korzyści płynące z wyboru reagentów i podejść, których celem finalnym jest opracowanie nowych metod funkcjonalizacji oligonukleotydów na podłożach stałych lub w roztworze. Wyniki prac nad reakcjami transestryfikacji zostały opublikowane132,136-143 w latach 1994 - 1996.
Zdaję sobie sprawę, że przedstawione w tej części rozprawy wyniki badań nad transestryfikacją i aminolizą diestrów H-fosfonianowych nie wyczerpują zagadnienia, ale, moim zdaniem, dostarczyły one istotnych danych jakościowych i ilościowych, które mogą być pomocne w dalszych pracach.
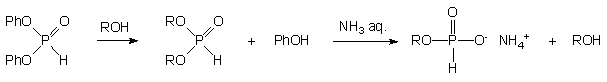
Mając na uwadze opisaną w poprzednich rozdziałach niniejszej rozprawy szczególną podatność arylowych diestrów H-fosfonianowych na transestryfikację można zakładać, że handlowo dostępny fosforyn difenylowy w reakcji z nadmiarem alkoholi będzie ulegał podstawieniu do H-fosfonianów dialkilowych, które następnie, hydrolizowane w odpowiednich warunkach, powinny tworzyć alkilowe monoestry typu 171 (Schemat 40). Przeprowadzony według tej koncepcji eksperyment, w którym fosforyn difenylowy traktowano 2-cyjanoetanolem (2 ekw.) w pirydynie prowadził w szybkiej reakcji (< 3 min.) do symetrycznego dialkilowego H-fosfonianu 179 (31P NMR). Diester 179 bez izolacji poddano hydrolizie wodnym roztworem amoniaku (10 min.) do H-fosfonianu 2-cyjanoetylowego 180 jako jedynego produktu. Stosując proste i szybkie procedury, związek ten izolowano z wysokimi wydajnościami w formie krystalicznej. Powyższe podejście, w przypadku wielu innych alkoholi dawało równie dobre rezultaty i zostało opublikowane w 1996 r. jako ogólna metoda syntezy różnych H-fosfonianowych monoestrów alkilowych.86
 |
122
124 |

|
|
|
| d: X = -(CH2)5-
e: X = -(CH2)6- f: X = -(CH2)2O(CH2)2- |
Schemat 41
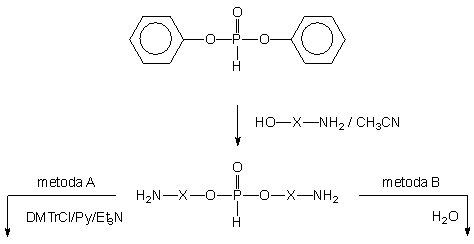
W tym przypadku szczególnie wyraźnie ujawnił się wpływ środowiska reakcji na jej chemoselektywność. Reakcje transestryfikacji fosforynu difenylowego aminoalkoholami (2 ekw.) w pirydynie prowadziły do mieszaniny produktów, w której oprócz pożądanych diestrów typu 165 obecne były (31P NMR) związki zawierające wiązania P-N (od 6% w przypadku aminoetanolu 124a do 45% w przypadku 6-aminoheksanolu 124e; Tabela 11). Natomiast ta sama reakcja w acetonitrylu prowadziła wyłącznie do fosforoestrowych produktów typu 165, bez różnicy, który aminoalkohol używano do transestryfikacji. Jest prawdopodobne, że przechodzenie ze środowiska zasadowego (pirydyna) do obojętnego (acetonitryl) przesuwa dyskutowaną wcześniej, równowagę form tautomerycznych aminoalkoholu na tyle mocno w kierunku alkoksylowego jonu obojnaczego, że reakcja transestryfikacji jest zdecydowanie dominująca.
Tabela 11. Produkty reakcji aminoalkoholi z fosforynem difenylowym w pirydynie
|
alkohol |
n |
(dP ~8 - 9 ppm) [%] |
(dP ~13 - 15 ppm) [%] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
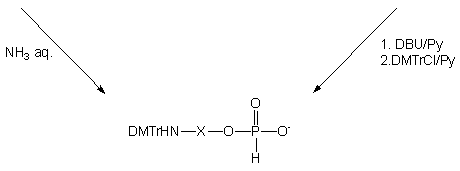
Ponieważ symetryczne diestry typu 165 były jedynymi zawierającymi fosfor produktami transestryfikacji prowadzonej w acetonitrylu, podjąłem próby wprowadzenia in situ ochronnej grupy dimetoksytrytylowej na funkcję aminową (Schemat 41, metoda A). Reakcje prowadziłem wobec dodanej pirydyny (1 : 1 v/v) i trietyloaminy (4 ekw.), stosując nieznaczny nadmiar (1,1 ekw. na grupę aminową) chlorku dimetoksytrytylu. Po 16 h N-trytylowany symetryczny diester typu 181 zhydrolizowałem (wodny 32% roztwór amoniaku - 1/2 objętości mieszaniny reakcyjnej) do pożądanego monoestru typu 182. Pomimo, iż reakcje zachodziły niemalże ilościowo (31P NMR), wydajności końcowych produktów typu 182, izolowanych chromatograficznie były raczej niskie (40 - 50%).
Niezadowalające wydajności były najprawdopodobniej spowodowane ograniczoną trwałością grupy N-dimetoksytrytylowej w obecności nadmiaru fenolu, w pierwszych fazach chromatografii. Dlatego też podjąłem próby syntezy syntonu nienukleotydowego 182, zmieniając kolejność poszczególnych etapów (metoda B). Produkt transestryfikacji fosforynu difenylowego, symetryczny H-fosfonian di(aminoalkilowy) 165, hydrolizowałem, dodając do mieszaniny reakcyjnej niewielką ilość wody (3 ekw.). Reakcja hydrolizy zachodziła wolno (120 h), ale w miarę jej postępu aminoalkilowe H-fosfoniany typu 173 krystalizowały z mieszaniny reakcyjnej z wysokimi wydajnościami (173d - 79%, 173e - 71% i 173f - 83%). Wprowadzanie grupy dimetoksytrytylowej na funkcję aminową aminoalkilo-H-fosfonianów typu 173 było trudne ze względu na ich bardzo słabą rozpuszczalność w pirydynie (a także innych rozpuszczalnikach organicznych), związaną, jak sądzę, z występowaniem tych związków w formie jonu obojnaczego. Dodanie do zawiesiny aminoalkilo-H-fosfonianu 173d - f w pirydynie, silnej zasady - DBU, w celu przekształcenia jonu obojnaczego w formę soli [DBU-H+][aminoalkil-O-P(O)HO-] radykalnie poprawiło rozpuszczalność substratu, tak że reakcja N-trytylowania zachodziła w układzie homogennym. Produkty 182d - f po standardowym przerobie izolowano na kolumnie wypełnionej żelem krzemionkowym z wydajnościami odpowiednio 81, 74 i 89%. Związki 173d - f oraz ich pochodne 182d - f scharakteryzowano metodami spektroskopowymi (1H i 31P NMR) i chromatograficznymi (TLC).
Powyżej opisaną metodę syntezy pochodnych aminoalkoholi (opublikowaną w 1996 roku),145 angażującą tanie, komercyjne reagenty mogę polecać jako prostą i wydajną metodę syntezy H-fosfonianowych syntonów nienukleotydowych typu 182 o właściwościach chemicznych umożliwiających ich włączanie w regularne cykle syntetyczne metody H-fosfonianowej otrzymywania oligonukleotydów.
