
Modyfikacje w pozycjach 1', 2' i 4' szkieletu cukrowego
Idea modyfikacji reszty cukrowej (C-1', C-4' i 2'-OH) oparta jest na założeniu,
że reszta ta nie jest bezpośrednio uwikłana w proces hybrydyzacji, w związku
z czym jej modyfikacje nie powinny w istotny sposób wpływać na trwałość
dupleksu. Wybór takiego sposobu modyfikacji z reguły wymaga przygotowania
w pierwszej kolejności odpowiednio zmodyfikowanego nukleozydu, następnie
jego amidofosforynu lub H-fosfonianu i wprowadzenie go do oligomeru. Syntony
tego typu można wprowadzać w dowolnej pozycji łańcucha oligonukleotydowego.
Możliwe jest też wprowadzanie do oligomeru wielu tego typu jednostek.
Modyfikacje pozycji 1'
Amidofosforyn nukleozydu zmodyfikowanego w pozycji 1' (85)88
otrzymany został w wieloetapowej syntezie (Schemat 11), w której substratem
był 2,3'-O-anhydro-1-(1,4,6-tri-O-benzoilo-b-d-fruktofuranozylo)uracyl.
Po hydrolizie mostka tlenowego 2,3' grupa 2'-OH usunięta została działaniem
N,N'-tiokarbonyloimidazolu i wodorkiem tributylocyny wobec 2,2'-azobis(izobutyronitrylu).
Benzoilowe grupy ochronne tak otrzymanej 3'-deoksypsikourydyny usunięto
następnie roztworem NaOH. Reszty -OH w pozycjach
4' i 6' zablokowano dwufunkcyjną grupą 1,1,3,3-tetraizopropylodisiloksan-1,3-diylową
(TIPDS), a w pozycji 1'-OH przyłączono diaminę za pośrednictwem N,N'-karbonylodiimidazolu.
Wolną resztę -NH2 zablokowano grupą
9-fluorenylometoksykarbonylową (Fmoc), a resztę siloksanową usunięto z
pozycji 3'-O i 5'-O działaniem fluorku tetrabutyloamoniowego. Ostatecznie
w grupę 5'-hydroksylową zablokowano dimetoksytrytylem i przeprowadzono
fosfitylację w pozycji 3'-O, co doprowadziło do syntonu 85.
Włączany był on do oligonukleotydów wg standardowych procedur, a po odblokowaniu
i oczyszczeniu oligomeru reszta 1'-aminoalkilowa wykorzystana była do przyłączenia
pirenu i antrachinonu.
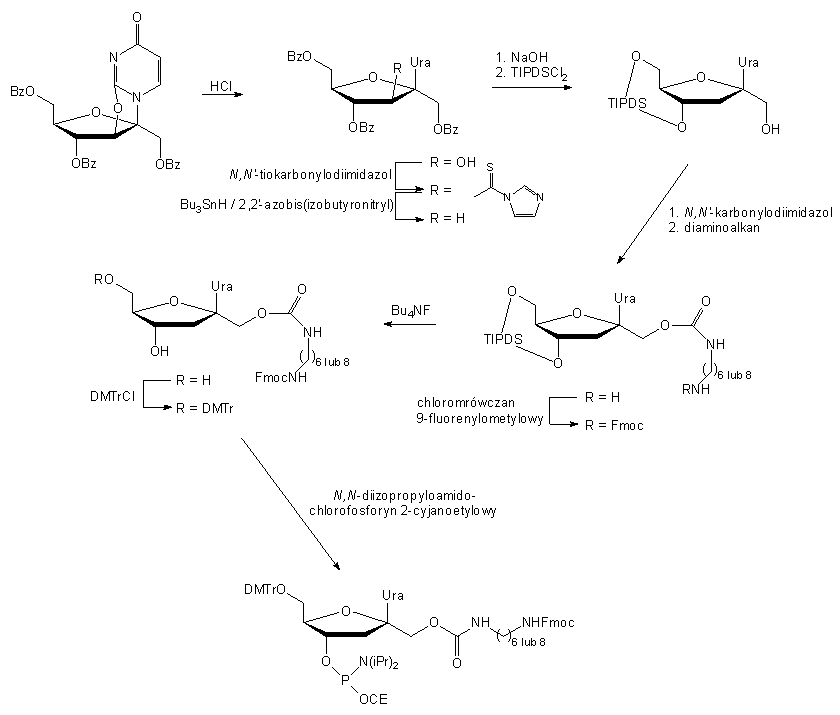
85
Schemat 11
W innym podejściu61, stosując amidofosforyn typu 86,
wprowadzono w wybrane pozycje łańcucha oligonukleotydowego jednostki 3'-deoksypsikotymidynylowe
z pozycją 1'-hydroksylową zabezpieczoną resztą lewulinylową.
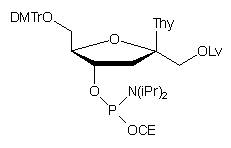
86
Po zakończeniu syntezy oligomeru grupę lewulinylową usunięto selektywnie
octanem hydrazyny i do uwolnionej reszty 1'-OH przyłączono amidofosforyn
typu 57 (*) niosący resztę glikolanu
trifluoroetylowego lub chloroetylowego jako miejsce dalszej modyfikacji.
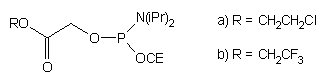
57
Tego typu ugrupowanie karboksyestrowe pod wpływem diaminy przekształca
się łatwo w ugrupowanie aminoalkilokarbamidowe (związek typu 87).
Po usunięciu wszystkich grup blokujących, w reakcji z chlorkiem 4-(dimetyloamino)azobenzeno-4'-sulfurylu,
do reszty aminoalkilowej w pozycji 1' przyłączono grupę dabsylową jako
znacznik nieradioizotopowy sondy.
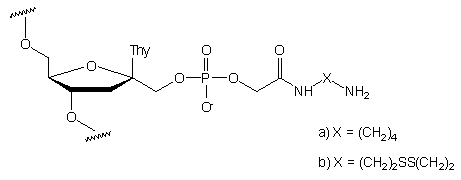
87
Modyfikacje pozycji 4'
Modelowanie komputerowe wykazało, że przyłączenie łańcucha alkilowego do
węgla 4' ma znacznie mniejszy wpływ na stabilność dupleksu DNA-DNA niż
podobne modyfikacje w innych pozycjach reszty cukrowej. Weryfikację eksperymentalną
obliczeń i wyników komputerowych zrealizowano przez syntezę oligomeru,
w którym w wybranych pozycjach ulokowano odpowiednio modyfikowane jednostki
nukleotydowe89. W tym celu przed syntezą oligonukleotydu konieczne
było przygotowanie odpowiednio modyfikowanego amidofosforynu 88.
Substratem do syntezy tego ostatniego był unikalny nukleozyd (4'- hydroksy-2'-deoksyurydyna),
który w siedmiu etapach syntetycznych przekształcono w 4'-aminoalkilo-2'-deoksyurydunę
i, po zablokowaniu funkcji 5'-OH, w amidofosforyn 88.
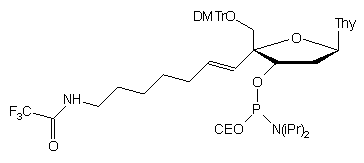
88
Przeprowadzono syntezy szeregu oligomerów z modyfikacjami w różnych
pozycjach sekwencji. Do odsłoniętych w trakcie końcowego odblokowania grup
aminoalkilowych przyłączano biotynę, otrzymując zestaw sond molekularnych
modyfikowanych w jednej lub kilku pozycjach. Porównawcze pomiary efektywności
hybrydyzacji wykazały, że każdy 4'-modyfikowany nukleotyd obniżał Tm
jedynie o 1,2 - 1,7°C, co potwierdziło eksperymentalnie wcześniejsze wyniki
otrzymane z modelowania komputerowego.
Modyfikacje pozycji 2'-OH
W odróżnieniu od modyfikacji w pozycjach 1' i 4', które wymagały syntezy
nienaturalnych jednostek nukleozydowych jako substratów, do modyfikacji
w pozycji 2' wykorzystano naturalne rybonukleozydy, a reakcje funkcjonalizacji
ukierunkowano na alkilowanie grupy 2'-hydroksylowej. Przykładem takiego
podejścia może być alkilowanie bromkiem S-trytyloalkilowym odpowiednio
przygotowanego nukleozydu z wolną grupą 2'-OH90. Po fosforynacji
otrzymywano amidofosforyn modyfikowanego nukleozydu 89, który
włączano do oligonukleotydu.
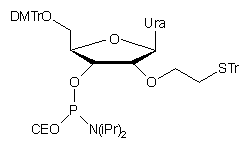
89
Ze względu na obecność objętościowej grupy trytylowej w sąsiedztwie
centrum fosforowego, po to, by zapewnić dostatecznie wysoką wydajność przyłączenia
tak modyfikowanego syntonu do łańcucha oligonukleotydowego, konieczne było
wydłużenie czasu kondensacji z 1 min. do 10 min. Próby zastąpienia grupy
trytylowej mniej objętościowymi grupami tert-butylową czy 2-cyjanoetylową
zakończyły się niepowodzeniem.
W podobny sposób otrzymano analogicznie zmodyfikowany amidofosforyn
nukleozydu 9091. Zastosowanie dłuższego łącznika
n-heksylenowego w miejsce etylenowego nie zmieniło zasadniczo efektywności
kondensacji i także w tym przypadku konieczne było wydłużenie czasu kondensacji
do 10 min.
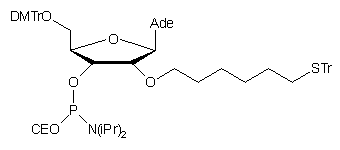
90
W obu podejściach należało stosować na etapie utleniania wodoronadtlenek
tert-butylowy w miejsce standardowo używanego I2/H2O,
gdyż w obecności jodu grupa trytylotioeterowa była nietrwała. Z oligomeru,
po syntezie usuwano grupę trytylową (AgNO3), a do wygenerowanej
reszty -SH przyłączano piren, fluoresceinę czy fenantrolinę jako grupy
reporterowe.
Ponieważ alkilowanie nukleozydów wiąże się z niebezpieczeństwem modyfikacji
reszt zasadowych, T. H. Keller i R. Haner zaproponowali przeprowadzenie
reakcji alkilowania odpowiednio blokowanej rybozy [3,5-O-(1,1,3,3-tetraizopropylodisiloksan-1,3-diylo)-b-d-rybofuranozydu
metylowego] i dopiero tak modyfikowany cukier wykorzystywano w reakcji
glikozylacji tyminy lub adeniny92. Uzyskany rybozyd tyminy posłużył
do syntezy pochodnej cytydynowej, nie przedstawiono natomiast metody otrzymywania
modyfikowanej w ten sposób guanozyny. Dimetoksytrytylowanie i fosforynowanie
tak otrzymanych nukleozydów prowadziło do amidofosforynów typu 91a
- c, niosących w pozycji 2'-O różne grupy modyfikujące93.
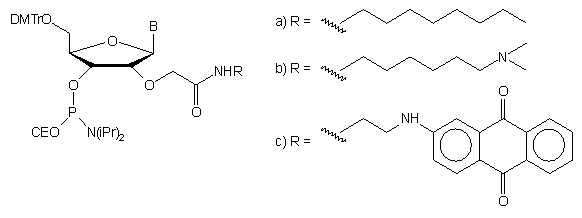
91
Otrzymane oligonukleotydy pozwoliły porównać wpływ charakteru reszty
modyfikującej na trwałość dupleksu DNA-DNA. I tak, grupy lipofilowe w pozycji
2'-O (np. n-oktyl) powodowały destabilizację dupleksu (obniżenie
Tm), natomiast grupy hydrofilowe (kation n-alkiloamoniowy)
czy interkalujące (antrachinon) wywierały efekty stabilizujące dupleks.
Wprawdzie prace te nie opisywały syntezy sond molekularnych z nieradioizotopowymi
grupami reporterowymi, jednak uważam, że mieszczą się w tematyce niniejszego
przeglądu.
Podsumowanie
Opisane powyżej metody modyfikacji pozycji 1', 4' i 2'-OH pozwalają na
wprowadzanie różnych ugrupowań w dowolne miejsce oligonukleotydu. Modyfikacje
te nie powodują zwykle znaczącego pogorszenia właściwości hybrydyzacyjnych
oligomerów, a nawet mogą je polepszyć. Umożliwiają przeprowadzanie różnorodnych
badań oddziaływań DNA - DNA i DNA - białko. Pomimo faktu, że molekularne
sondy oligonukleotydowe opisane w tym rozdziale mają pewne zalety w porównaniu
z sondami o innym typie modyfikacji, to zalety te nie są aż tak dominujące,
by polecać modyfikacje w pozycjach 1', 4' czy 2'-O jako zdecydowanie najlepsze.
Dodatkowo, ze względu na stopień trudności i złożoność przygotowania tego
typu sond, ich zastosowania będą ograniczone jedynie do specjalnych celów
lub przypadków, gdy inne typy modyfikacji nie będą możliwe.
