|
Nr
|
Zasada
|
Komentarz
|
Lit.
|
|
92
|
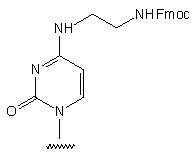
|
-
Otrzymywanie: transaminacja cytydyny etylenodiaminą, następnie wprowadzenie
grupy Fmoc i standardowe reakcje prowadzące do przyłączenia nukleozydu
do podłoża stałego.
-
Odblokowanie grupy aminowej podczas końcowego działania amoniakiem.
-
Do oligomeru po odblokowaniu przyłączono enzym (AP) poprzez dodatkowy łącznik
z wolną grupą tiolową (str. *).
|
94
|
|
93
|
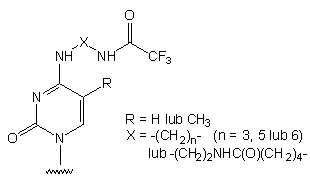
|
-
Otrzymywanie nukleozydu: z 5',3'-O-(tetraizopropylodisiloksano-1,3-diylo)-4-N-tosylocytydyny
w 4 etapach z wyd. 46%95; z 5'-dimetoksytrytylotymidyny poprzez
pochodną 4-triazolopirymidynową, 4 etapy, wyd. 54%18;z cytydyny
przez pochodną 4-O-(2-nitrofenylową) w 6 etapach z wyd. 41%96;
z cytydyny przez transaminację w 3 etapach z wyd. 33%97.
-
Odblokowanie grupy aminowej podczas końcowego działania amoniakiem.
|
18,
95-97
|
|
94
|
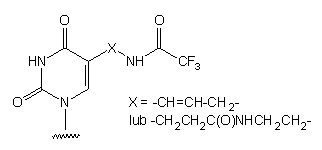
|
-
Otrzymywanie nukleozydu: z 5-chlorortęcio-2'-deoksyurydyny w 2 etapach
z wyd. 21%98 lub w 3 etapach z wyd. <50%97.
-
Odblokowanie grupy aminowej podczas końcowego działania amoniakiem.
|
97, 98
|
|
95
|
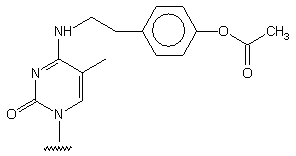
|
-
Otrzymywanie nukleozydu: z 4-tiotymidyny, proces trójetapowy, wyd. 34%.
-
Resztę fenolową wykorzystano po syntezie oligomerów do wprowadzania atomów
125I. Wydajność jodowania wynosiła ok. 10%.
|
99
|
|
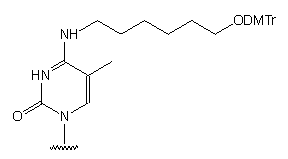
96
|
|
-
Otrzymywanie nukleozydu: z 5'-dimetoksytrytylotymidyny, w 4 etapach, wyd.
53%100 lub z 5'-dimetoksytrytylo-4-tiotymidyny, w 2 etapach,
wyd. 79%62.
-
Związek 96 służy do uzyskiwania rozgałęzionych oligonukleotydów
(typu "drzewo") umożliwiających wprowadzanie wielu grup reporterowych.
|
62, 100
|
|
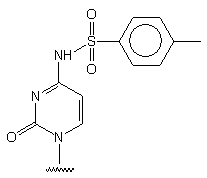
97
|
|
-
Otrzymywanie nukleozydu: z cytydyny, w dwóch etapach, wyd. 74%.
-
Aby uzyskać wolne reszty aminowe, oligomer, po odcięciu od podłoża stałego
(NH3 aq. stęż., 1h, temp. pok.), inkubowano w 1,2-bis(2-aminoetoksy)etanie*
(80°C przez noc). Pod wpływem tej diaminy reszty 4-N-p-tosylowe
ulegały transaminacji, a jednocześnie następowało całkowite odblokowanie
oligomeru.
|
101
|
|
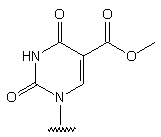
98
|
|
-
Otrzymywanie nukleozydu nietrytylowanego: z 5-jodo-2'-deoksyurydyny, w
jednym etapie, wyd. 95%.
-
Oligomery na podłożu poddawano działaniu diamin, które reagowały z ugrupowaniem
estrowym 5-metoksykarbonylouracylu dając pochodne z wolnymi grupami aminowymi.
Równocześnie następowało całkowite odblokowanie i odcięcie oligomeru od
podłoża stałego.
|
102
|
|
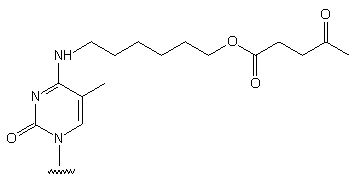
99
|
|
-
Otrzymywanie nukleozydu: z 5'-dimetoksytrytylotymidyny, w 4 etapach, wyd.
56%.
-
Po wprowadzeniu kilku jednostek nukleozydowych typu 99 funkcję
hydroksylową odblokowywano hydrazyną i prowadzono dalej syntezę, uzyskując
modyfikowany oligomer typu "grzebień".
|
100, 103, 104
|
|
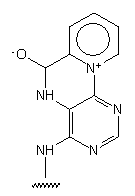
100
|
|
-
Otrzymywanie nukleozydu: z inozyny, w trzech etapach. Kluczowa reakcja
to fotochemiczne przekształcenie chlorku N-(3',5'-di-O-acetylo-2'-deoksynebularyn-6-ylowo)
- pirydyniowego w diacetylową pochodną luminarozyny (wyd. 30%).
-
Deoksyluminarozyna jest silnym fluoroforem.
|
105
|
|
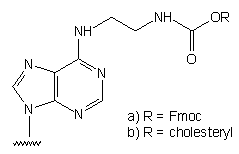
101
|
|
-
Otrzymywanie nukleozydu: z rybozydu 6-chloropuryny, w 3 etapach, wyd. 66%
(101a) i 63% (101b).
-
Nukleozydy typu 101 przyłączono do podłoża stałego i wykorzystano
do syntezy oligomerów modyfikowanych na końcu 3'.
|
106
|
|
102
|
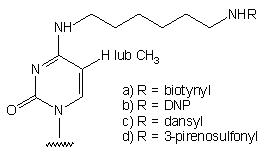
|
-
Otrzymywanie nukleozydów: z 3',5'-di-O-acetylo-2'-deoksyurydyny (lub tymidyny)
poprzez pochodną 4-tio, w 4 etapach, wyd. rzędu 40%.
-
Grupa 2,4-dinitrofenylowa okazała się nietrwała w warunkach odblokowania
(30% rozkładu po 16h w NH3 aq. stęż., 55°C).
|
78
|
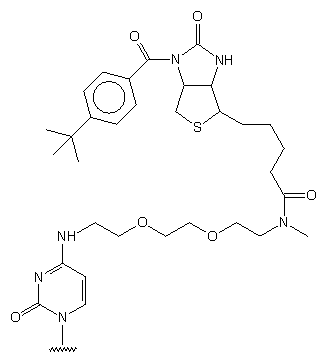
103
|
|
-
Otrzymywanie nukleozydu: z 2'-deoksyurydyny, w 7 etapach, wyd. 16%.
-
Jako łącznik wykorzystano 1,2-bis(2-metyloaminoetoksy)etan ze względu na
jego podatność na hydratację (przypis str. *).
-
Pozycja N-1 pierścienia ureidowego biotyny chroniona była grupą p-(tert-butylo)benzoilową.
Związek 103 włączano w dowolne miejsce oligonukleotydu.
|
107
|
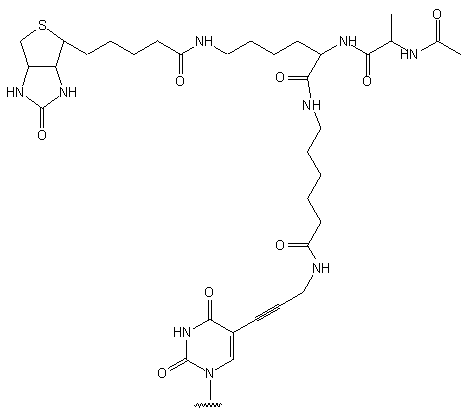
104
|
|
-
Otrzymywanie: wychodząc z 5-jodo-2'-deoksyurydyny, w 3 etapach otrzymano
pochodną 5-[3-(Boc-amino)prop-1-yn-1-ylową lub 5-[3-(Pix-amino)prop-1-yn-1-ylową
z wydajnością odpowiednio 33 i 53%. Pochodne te przyłączono do podłoża
stałego i w 5 etapach przeprowadzono w podłoże 104, zawierające
pochodną deoksyurydyny, wykorzystując reakcje i odczynniki stosowane w
syntezie oligopeptydów. Podłoże to wykorzystano następnie do standardowej
syntezy oligonukleotydów.
-
Biotynylowany 23-mer uzyskano z niską wydajnością (2%).
-
Autorzy nie badali możliwości zachodzenia reakcji ubocznych w ureidowym
pierścieniu biotyny podczas syntezy oligonukleotydu.
|
108
|
|
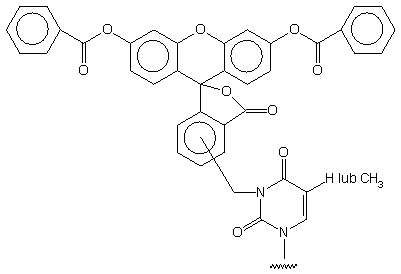
105
|
|
-
Otrzymywanie nukleozydu: z 5'-O-dimetoksytrytylotymidyny, w 2 etapach,
wyd. 27%. Do alkilowania pozycji N-3 użyto 3',6'-dibenzoilo-4(5)-(bromometylo)fluoresceiny
otrzymanej z bezwodnika 4-metyloftalowego i rezorcyny (3 etapy, 50%).
-
Z nukleozydu otrzymano 5'-O-amidofosforyn i przyłączono do oligonukleotydu
z wydajnością ok. 70% uzyskując wiązanie 5' - 5'.
-
Ochronne grupy benzoilowe usunięto w trakcie działania amoniakiem. Wiązanie
fluoresceiny z nukleozydem okazało się stabilne w standardowych warunkach
odblokowania.
|
109
|
|
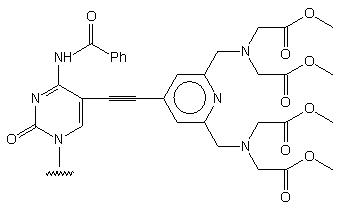
106
|
|
-
Otrzymywanie nukleozydu: z N4-benzoilo-5-jodo-2'-deoksycytydyny
lub
5-jodo-2'-deoksyurydyny, w 2 etapach, wydajności: odpowiednio 60% i
81%.
-
Przed końcowym działaniem amoniakiem należy zhydrolizować wiązania estrowe
roztworem NaOH.
-
Ugrupowanie typu 106 wykorzystano do chelatowania kationów
Eu3+ i Tb3+, co pozwoliło na zastosowanie szybkiej
fluorymetrii do wykrywania tak zmodyfikowanych oligomerów.
|
110
|
|
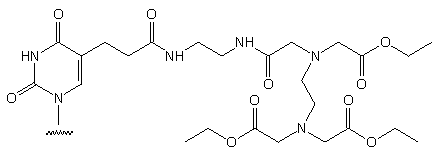
107
|
|
-
Otrzymywanie nukleozydu: z 2'-deoksyurydyny, w 4 etapach, wyd. 31%.
-
Do odblokowania użyto 0,1 M NaOH zamiast amoniaku.
-
Pochodną typu 107 wykorzystano do chelatowania kationów Fe2+,
które katalizowały specyficzną hydrolizę komplementarnej nici DNA w sąsiedztwie
modyfikacji.
|
37
|

